Spanish Front sight for two - how it affects libido in women and men
Contents Dietary supplement based on an extract obtained from the Spanish beetle (or Spanish beetle...
Cystic fibrosis is a severe hereditary disease. It is one of the five most common genetic pathologies. This is an incurable disease. The life expectancy of patients with cystic fibrosis is significantly shorter than that of healthy people. And the only chance to prevent this disease today is a genetic examination of both future parents. Let's talk about how to competently approach pregnancy planning, diagnostic methods and the disease itself. The material was prepared with the participation of experts - leading geneticists at the Next Generation Clinic.
On the left is a picture of the lungs of a healthy person, on the right is a picture of the lungs of a patient with cystic fibrosis
Cystic fibrosis, in Western countries the more common name is cystic fibrosis, which reflects the pathological changes that occur in the pancreas of patients - this is a severe and common hereditary disease. It cannot be treated and the only way to avoid it is to undergo genetic diagnosis at the pregnancy planning stage.
Why is cystic fibrosis dangerous?
With this disease, the functioning of the exocrine glands, which are also called exocrine glands, is disrupted. They are responsible for the production of secretions such as mucus, sweat, digestive juice, tears, and so on, which perform many functions in the body, for example, promote digestion, remove bacteria from the respiratory tract, and control heat exchange.
This is the algorithm that is relevant for the functioning of the exocrine glands of a healthy person. However, in patients with cystic fibrosis, their function is impaired and the mucus produced is too viscous, which leads to blockage of the exocrine ducts and creates so-called mucus plugs. As a result, the functioning of organs and entire systems of the body is disrupted. Most often, the human respiratory and digestive system suffers from this.
What is the cause of malfunction of the exocrine glands?
The cause of the breakdown is an abnormal gene that controls the membrane transport of substances such as salt and water, that is, their passage from one cell of the body to another. This failure causes the glands to produce mucus with insufficient amount of water - viscous. Hence, by the way, the name of the disease, formed from two Latin terms “mucus” and “viscous”. Thus, “cystic fibrosis” can be translated as “sticky mucus.”
The genetic nature of cystic fibrosis, the history of the discovery of mutations, the occurrence of mutations
Until the 20th century, scientists and doctors did not classify cystic fibrosis as an independent disease. When this did happen, for a long time it was classified as a childhood disease, because without proper drug therapy, the life of a patient with cystic fibrosis was cut short at an early age.
Due to late diagnosis and lack of understanding of the essence of the disease among doctors and
In the mid-20th century, doctors came to the conclusion that the disease was hereditary. It was possible to understand the root cause in the late 80s, when the gene for the transmembrane regulator of cystic fibrosis was discovered.
The cystic fibrosis transmembrane regulator gene, commonly known in medical circles as CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), is a gene that encodes the functioning of a protein under the same name - the cystic fibrosis transmembrane regulator.
This protein forms special channels in cell membranes (membranes), which are necessary so that the particles of chlorine ions included in the salt solution can pass from one cell to another. For its role in water-salt movement, this protein is called a channel protein. The CFTR gene is responsible for its work. And if a mutation occurs in it, then it makes adjustments to the functioning of the protein. Thus, the movement of water and salt from cell to cell is disrupted. This most severely affects the functioning of the exocrine glands, causing a disease such as cystic fibrosis.
What is a gene mutation?
This means that a small section of the gene is “lost,” or better yet, “falls out.” For this reason, the protein molecule also lacks part of the required fragment. The modified molecules cannot reach the cell membrane and form a channel for transporting chlorine ions.
However, depending on the mutations, the protein can behave differently. For example, it can reach the cell surface, but due to improper operation, never create a channel. The course of cystic fibrosis and its symptoms depend on which particular mutation occurred.
To date, the number of CFTR gene mutations discovered by scientists is almost two thousand. Among them, 10-12 are the most common. More often than others, according to statistics, there is a mutation that is designated as F508del. It is diagnosed in more than half of Russians who have or are carriers of cystic fibrosis.
At the same time, carriers may not even be aware of the presence of an abnormal gene, lead a healthy lifestyle and even be excellent athletes. Clinically, carriage does not manifest itself in any way and exists in the human body completely asymptomatically, but carries the risk of giving birth to a child with cystic fibrosis. Therefore, it is extremely important to take care of the health of your unborn child before pregnancy and undergo genetic testing for cystic fibrosis.
Laws of inheritance of cystic fibrosis. Parents are carriers of mutations.
Genes are the basic unit of heredity. They are located in chromosomes, which are located in the nucleus of the cells of our body. Moreover, the number of genes on one chromosome is thousands. Chromosomes are united in pairs - there are 23 of them in a healthy person. Genes, as a rule, are also found in pairs. Moreover, they can be the same, or they can differ - one copy can be healthy, and the second has a mutation.
At the time of conception, the child receives half the set of chromosomes from each parent - which means that it includes one chromosome from each pair, and therefore one gene from each pair of genes. So a child is born with his own individual paired set of chromosomes and genes. It is impossible to predict which genes from parental pairs will be passed on - healthy or defective.
If a child received a mutant gene from both parents, then with a 25% probability the child will be born with the terrible diagnosis of cystic fibrosis. There is an even greater chance - 50% - that the child, like his parents, will become a carrier, which means that the “sick” gene will continue to be inherited in this family.
It is important that carriers will not be aware of the terrible disease that can affect their children. Be healthy, active, free from pathologies and external manifestations of a latent mutant gene.
Due to the fact that carriers do not have any manifestations of cystic fibrosis, many are not even aware of this “family heritage”. And the disease is passed on from generation to generation. The carrier not only has no idea about the presence of the mutant gene, but also will not know that he may have already passed it on to his children.
This chain continues until a fatal meeting of two carriers occurs and during conception the child, with a probability of 25%, receives an abnormal gene from both parents. Then a child with cystic fibrosis is born into the family.
Therefore, it is so important to approach the birth of a child with full responsibility and understanding that hereditary diseases, and these are by far the most serious illnesses with which babies are born, can be genetically programmed even in people who appear to be absolutely healthy. And it doesn’t matter who they are - professional athletes, fans of extreme sports, promoters of healthy eating, or just people who care about their health, so that future children receive the best set of genes.
Cystic fibrosis is a disease that belongs to an autosomal recessive mode of inheritance.
What does it mean?
This means that the gene, the mutation of which leads to cystic fibrosis, is not located in the sex chromosomes, but in autosomes, and therefore can be inherited by both a boy and a girl.

Scheme of autosomal recessive inheritance, which includes cystic fibrosis
Clinical picture of the disease, examples of children
Cystic fibrosis is an incurable disease. The only way to prevent the transmission of a hereditary disease to children is to take a genetic test to determine if you are a carrier. This simple procedure - the analysis requires the venous blood of both future parents - can protect future children from a serious and terrible disease. Consultation with a geneticist should be a mandatory step in preparing for pregnancy.
What do children with cystic fibrosis have to contend with?
All drug therapy is aimed at maintaining the patient’s life. At the same time, cystic fibrosis is a catalyst for other diseases. If we talk about the most common clinical manifestations of cystic fibrosis, it is divided into three forms:
Cystic fibrosis also affects the reproductive system, which means that people with cystic fibrosis have problems conceiving.

A girl from America with pulmonary fibrosis. Her mother posted this photo on the Internet, accompanied by a story about her child’s daily courage and joint fight against the disease.
For a long time, the only way to make a diagnosis was sweat tests. Today, genetic research is increasingly being used. Since 2007, in our country, all newborns in the maternity hospital undergo mandatory neonatal screening for the five most severe hereditary diseases, including cystic fibrosis. The study can also be done during pregnancy. But we are only talking about an attempt at early diagnosis of cystic fibrosis, which, undoubtedly, is important for prescribing adequate therapy, but the disease itself does not go away. It is hereditary and incurable.
The life expectancy of people with cystic fibrosis varies greatly from country to country. So, in America today the average is 48 years. However, this is the exception rather than the rule. This was achieved thanks to titanic efforts and coordinated work of doctors, scientists and public organizations. They are also actively trying to fight cystic fibrosis in Russia. However, these efforts are not enough, as statistics confirm - in our country, patients with cystic fibrosis live on average 10-15 years less.

These are the kind of reminders you can find in American hospitals. They briefly provide all the important information about the disease - symptoms, as well as the treatment necessary for the patient.
What can be done today to prevent a child from being born with cystic fibrosis?
The only real way to prevent cystic fibrosis today is a genetic test for carrier status of hereditary diseases, which both future parents must undergo before conception. Only this will help to establish with maximum accuracy possible carrier status and find out whether the spouses are at risk of having a sick child.
Where to go for analysis?
Next Generation Clinic offers its patients a truly unique comprehensive study of genes for 21 hereditary diseases, including cystic fibrosis. The study itself is based on high-throughput sequencing technology – NextGen 21, which is recognized throughout the world as a “new word” in molecular genetic diagnostics. This technology allows thousands of DNA molecules to be sequenced simultaneously at high speed and large volumes of data. It allows you to find out your genetic status and assess the risk of having seriously ill children with the highest accuracy. In this case, all that is required from future parents is to donate venous blood.
At the same time, there is another diagnostic method - to take an analysis for frequent point mutations of the transmembrane regulator gene for cystic fibrosis. It is, of course, impossible to test a gene for all existing mutations. As you remember, there are about two thousand of them. But among them there are those that occur with high frequency. And today it is possible to test the gene for the presence of the most common abnormalities leading to cystic fibrosis.
Cystic fibrosis remains a mysterious disease for most Russians, and therefore raises many questions. Leading specialists at Next Generation Clinic answered those questions that they hear most often from their patients.
Could the child's illness have been prevented? What can I do to make sure my next child is healthy? Is it possible to prevent a child from getting sick if a mutation has been identified?
First, you need to come for a consultation with a geneticist. The second is to undergo the prescribed studies. The third step is to develop a plan for further action based on the test results together with a geneticist. What can couples do where both parents are carriers of cystic fibrosis, as well as families where the first child has already been born with this disease? Resort to preimplantation diagnostics, which allows you to select an embryo with a healthy gene, which will prevent inheritance of the disease. This is only possible if IVF technology is used. Another option is to use a donor who is not a carrier of cystic fibrosis.
Does the course of the disease depend on the mutation?
Yes. There is certainly a connection between the type of mutation and how severe cystic fibrosis will be. There are mutations in which the effect on the human body is more gentle. And vice versa. For example, if a baby at the time of conception was given abnormal genes from a parent with the same mutation, which doctors classify as “severe,” then the child will almost certainly have pancreatic insufficiency. But if one of the transmitted abnormal parental genes had a mutation in which the clinical picture of the disease is milder, then pancreatic insufficiency may not develop.
However, in each individual case, the disease will still proceed according to its own scenario. This is also indicated by the fact that in patients with the same gene mutation, the course of the disease may differ. The reason for this is the genetic characteristics of each patient and other non-genetic factors.
Why do a test if you can’t change the genes anyway?
This question shows the wrong attitude towards genetic diagnosis in general. It turns out this way - the spouses learn that there is a serious hereditary disease in their family, which can be passed on to future children, only after they have a sick child. And this is certainly not the best way to learn about genetic risks in this day and age. Modern technologies make it possible to take care of the health of future children in advance. And for all those who want to have a child, it is advisable to undergo testing for clinically significant mutations - those that doom them to disability, are bedridden, do not allow them to live a full life, or cause early death. These include cystic fibrosis.
If screening is done, does it need to be repeated or other tests done?
No. Abnormal genes that are responsible for hereditary diseases are something a person is born with and they cannot appear or disappear with age. As for other tests, everything will depend on the recommendations of a particular specialist. However, today genetic studies are the most accurate diagnostic method.
These are the questions that patients most often ask. And these are questions that geneticists themselves consider important to answer.
Does everyone need to undergo testing to look for mutations in the cystic fibrosis gene?
Preferably. This study, as well as studies for other severe hereditary diseases that have a recessive, that is, dormant principle of inheritance, is today recommended for all married couples planning children.
If the mutant gene is detected in both parents, then a choice arises: this could be preimplantation diagnosis of the fetus with the then obligatory IVF procedure, prenatal diagnosis (carried out during pregnancy) or the use of donor biomaterials (which also undergo genetic testing for carriage of severe monogenic diseases, such as cystic fibrosis).
Cystic fibrosis is a disease that, in the current Russian conditions, unfortunately, does not guarantee a long and fulfilling life for the patient. And the rule works here: it’s better to check once than not to check at all. Conducting research is a medical recommendation, but the recommendation is persistent.
Is it necessary to do a DNA test if there is no family history of this disease?
Preferably. Cystic fibrosis is a recessive disease, which means that it will only appear when two copies of the diseased gene - mom's and dad's - meet. Until this point, the abnormal gene may be inherited but not manifest clinically. Moreover, there may be no family history of cystic fibrosis patients, and the gene mutation will be a gift from a distant ancestor who was also a carrier.
We should not forget that sometimes it is very difficult to find traces of a hereditary disease in a family. Provided that even a century ago doctors themselves did not identify cystic fibrosis as an independent disease, and the first records of patients whose symptoms, as was already clear today, indicated cystic fibrosis, were much earlier.
Today, every clinically healthy person is a carrier of 10-15 mutations, without even knowing it. And at what point they may come together (the result of such a meeting is the birth of a sick child) cannot be predicted; the only way to prevent diseases such as cystic fibrosis is genetic research.
Do relatives need to do a DNA test if carrier status is determined in someone in the family?
Yes. Cystic fibrosis is a hereditary disease, which means that there is a high risk that immediate relatives, without knowing it, may also be carriers.
Cystic fibrosis is also called cystic fibrosis. This is a progressive disease of a genetic type. Because of it, an infection occurs in the lungs and gastrointestinal tract.
The function of the respiratory and gastric organs is limited. People with this condition have a defective gene that causes mucus to build up in the respiratory system, pancreas, or other organs.
The mucus in the lungs traps bacteria inside and prevents normal breathing. Thus, an infection constantly forms in the body of a healthy person, which leads to damage to the lungs, and respiratory failure may occur. If mucus is located in the pancreas, it prevents the formation of digestive enzymes that break down food in the stomach. Therefore, the body does not absorb vital nutrients.
The first symptoms of the disease were described in the forties of the 20th century. From the name it follows that “mukas” is a Greek root that means “mucus”, “viscus” is glue. If you put the two particles together, the disease can be translated literally as “mucous secretion.” It is secreted outward by various secretions of the body. The substance has high viscosity.
Doctors have clearly established that cystic fibrosis is a genetic disease. The disease is inherited from parents. Cystic fibrosis is not contagious, even if a person has harmful working conditions and a difficult lifestyle, he will not get sick. Doctors have discovered that the disease is not related to a person’s gender. Cystic fibrosis can affect both men and women.
The type of transmission of the disease is considered recessive, but not the main one. The disease is encrypted at the genetic level. If only one of the parents has unhealthy genes, then most likely the child will be healthy. According to statistics, a quarter of the heirs are healthy, and half contain the cystic fibrosis gene in their bodies, but it is located at the chromosomal level.
Approximately 6% of the adult population of the Earth has material from this gene in their bodies. If a child is born from parents who have distorted chromosomal information, then only in a quarter of cases the disease is transmitted to the baby. It is this type of transmission of the disease that is called recessive.
The disease is not related to a person's sex because the material is not found in sex genes. An equal number of sick boys and girls are born each year. No additional factors influence a person’s gender. It doesn’t matter how the pregnancy went, how healthy the mother or father is, or what their living conditions are. This disease is transmitted only genetically. In the nineties there were noted main signs of the disease:
Endocrine glands are organs that supply the blood with biologically functional elements called hormones. Thanks to them, physiological processes are regulated. Disease of the endocrine glands is a symptom of cystic fibrosis. Organs in the human body that are responsible for producing communication, the following:

These organs include the salivary glands and pancreas. They are responsible for the production of bronchial secretions. In adults, the symptoms of cystic fibrosis are the pathological thickness of the physiologically necessary mucous layer. Dense mucus forms in the lumen of the bronchial tree. Therefore, the respiratory organs are excluded from the life process. The body stops receiving the necessary oxygen, so pulmonary atelectasis forms.
Due to cystic fibrosis, a fatty and protein layer is formed and distorted in the liver, bile stagnation occurs, and as a result, the patient suffers from cirrhosis of the liver. The disease cystic fibrosis has another name - cystic fibrosis.
If a newborn baby has intestinal obstruction, the intestines are the first to suffer. This occurs due to swelling of the submucosal layer of the intestine. The disease is almost always accompanied by other gastrointestinal disorders.
Symptoms of the disease are detected in early childhood. Diagnosis of cystic fibrosis will help identify solutions and provide effective treatment. If symptoms are not detected early in life, they may occur later in life. How to tell if a person has cystic fibrosis:

The disease has clinical types; depending on the course, there are intestinal, atypical, meconium ileus, bronchopulmonary, and pulmonary forms. The disease has a genetic form and is closely related to the everyday physiological processes occurring in the body. Typically, clinical manifestations of cystic fibrosis are detected in a newborn child. There are cases when the disease was detected during intrauterine development of the fetus. Meconium ileus is often diagnosed in newborns.
Meconium is the name given to original feces. These are the first bowel movements of a newborn baby. If the child is healthy, then feces are released on the first day. In the disease, fecal retention is associated with the absence of a pancreatic enzyme called trypsin. The intestines do not form this element, and as a result, the feces stagnate. This occurs in the colon and cecum.
 As the disease progresses, symptoms appear:
As the disease progresses, symptoms appear:
During examination, the doctor may notice an increased vascular pattern on the tummy; when tapping, a drumming sound is detected. The child's mood often changes: at first he is restless, and then lethargic. He lacks the necessary motor activity. The skin is pale and dry. Due to the fact that the child does not excrete feces on time, the body becomes poisoned with products of internal decay. When listening to the heart, the following symptoms are revealed:
If a newborn is sick with cystic fibrosis, then he is diagnosed with swelling of the loops of the small intestine, and also a sharp decrease in the intestinal tract in the lower part of the tummy. Due to the fact that the child is too small, his condition is rapidly deteriorating. The baby may experience this as a complication.
It occurs due to rupture of the intestinal walls. A complication also occurs in the form of pneumonia; in newborns it occurs in a protracted and severe form.
If the patient has a pulmonary form of the disease, then he has pale skin and low weight. But at the same time the person has a good appetite. If a newborn has a disease, then already in the first days of life he develops a cough, the intensity of which constantly increases. Pertussis-like attacks begin, which are called reprise. How does lung damage occur?

Mucus forms in the patient's lungs, which is an excellent environment for the development of microorganisms that can cause pneumonia. The sputum subsequently becomes purulent and mucous, and streptococcus, pathogenic microorganisms and staphylococcus are released from it. Inflammation of the lungs occurs in a complex and severe form, usually caused by cystic fibrosis the following complications:
When a doctor listens to the lungs, moist rales are differentiated. The sound above the lungs has a box “echo”. The patient's skin is pale and dry.
 With a benign course of the disease, signs of the disease appear only in an adult. At this time, the body develops compensation mechanisms. Symptoms gradually increase, chronic pneumonia develops, and then pulmonary failure is diagnosed. Bronchitis gradually appears with the transition to pneumosclerosis.
With a benign course of the disease, signs of the disease appear only in an adult. At this time, the body develops compensation mechanisms. Symptoms gradually increase, chronic pneumonia develops, and then pulmonary failure is diagnosed. Bronchitis gradually appears with the transition to pneumosclerosis.
In cystic fibrosis, the upper respiratory tract is also involved. In addition to the disease, adenoids, appendages in the sinuses and proliferation of the nasal mucosa begin to form. A person may have chronic tonsillitis. The disease does not go unnoticed, the patient’s appearance changes:
In case of illness, the study will reveal thick mucus in the lumens of the small bronchi. Next, doctors will conduct an X-ray examination, which usually diagnoses a decrease in the branches of the small bronchi.
 In a healthy person, digestion proceeds normally due to the secretion of secret components necessary for this process. Digestive insufficiency is detected in patients with cystic fibrosis. This is due to the minimal production of the necessary fluids.
In a healthy person, digestion proceeds normally due to the secretion of secret components necessary for this process. Digestive insufficiency is detected in patients with cystic fibrosis. This is due to the minimal production of the necessary fluids.
Symptoms of the disease occur when a child stops drinking only breast milk and his diet becomes varied. In this case, digestion of food becomes more difficult, food does not move through the gastrointestinal tract. Next, active putrefactive processes occur.
Externally, the disease manifests itself as bloating and frequent bowel movements. At the same time, the child’s appetite does not decrease; he consumes more food than a healthy baby. But weight gain does not occur, while muscle tone is reduced, the skin is inelastic and flabby. A person with cystic fibrosis produces a minimal amount of saliva, so food is washed down with plenty of liquid. Dry food becomes very difficult to chew. The pancreas does not have the necessary secretion, so the child is often diagnosed with diabetes mellitus, gastric ulcers and abnormalities of the digestive tract.
The stomach does not absorb substances necessary for life, so the body feels a lack of vitamins. The patient may develop hypovitaminosis. In infants, due to a lack of proteins in the plasma, swelling is observed. The liver also suffers, a large accumulation of bile is found, which leads to the formation of cholestasis. Externally, this disease is characterized by an increase in the size of the liver, dry skin, and the skin acquires a yellowish tint.
This form is considered one of the most complex types. Already from the first days, the newborn develops signs of intestinal and pulmonary form of cystic fibrosis:

Mixed forms of the disease are directly related to the patient's age. This makes the disease more pronounced and malignant. The younger the child is, the worse the prognosis for relieving signs of the disease.
If the patient experiences weight loss, the disease is characterized by hypertrophy. Usually the patient lags behind in physical development. Diseases of the bronchi, sinuses, and lungs are also observed, and respiratory failure develops. A common symptom of cystic fibrosis is pancreatitis and dyspeptic complaints. To accurately identify the disease, laboratory and clinical studies are carried out. These include:

The first test that is performed is a sweat test. It is sampled three times; the liquid is collected after provocative electrophoresis. Coprological studies are done to determine chymotrypsin in the stool. If pancreatic insufficiency is detected, the analysis will give a result of more than 25 moles per day. The presence of chymotrypsin is determined using a variety of tests.
The most accurate method for determining the disease is DNA diagnostics. Now it is widely used by doctors, but this method has several significant disadvantages: In sparsely populated regions, the method is usually unavailable. DNA diagnostics are expensive. Doctors also use perinatal diagnostics. To determine the history, amniotic fluid is taken. Analysis is possible after 20 weeks. The error of the result varies within no more than 4%.
All actions to treat the disease are symptomatic. Treatment of cystic fibrosis is aimed at improving the patient's condition. The main thing in therapy is the restoration of nutrients in the gastrointestinal tract. Patients have poor digestion, so their diet should be 30% more fortified and saturated than the usual diet of a healthy person.
 The main diet is to consume the required amount of protein. The patient must include meat products, eggs, fish and cottage cheese in the diet. At the same time, you need to reduce your consumption of fatty foods to a minimum. It is forbidden to eat beef and pork, because meat has refractory fats. The lack of fatty acids is compensated by the consumption of polyunsaturated fatty compounds. Pancreatic juice and lipase are not needed to break down these elements. Usually it is these substances that the patient’s body lacks.
The main diet is to consume the required amount of protein. The patient must include meat products, eggs, fish and cottage cheese in the diet. At the same time, you need to reduce your consumption of fatty foods to a minimum. It is forbidden to eat beef and pork, because meat has refractory fats. The lack of fatty acids is compensated by the consumption of polyunsaturated fatty compounds. Pancreatic juice and lipase are not needed to break down these elements. Usually it is these substances that the patient’s body lacks.
Doctors recommend Minimize lactose and carbohydrate intake. Analyzes determine what kind of sucrose deficiency the patient has. Lactose is classified as milk sugar, which is found in dairy products. The patient's pancreatic juice contains a deficiency of the enzyme that is responsible for the breakdown of food. Therefore, dairy products will lead to poor digestion.
In the summer, a person's sweating increases, and accordingly, there is a deficiency of sodium chloride in the body. Its deficiency is compensated by adding the substance to food. A person with cystic fibrosis should have plenty of fluids in their daily diet, as well as foods that include vitamins of all groups and nutrients. You need to consume butter in the required quantity. The menu should also include fruits and vegetables.
Due to disruptions in the digestive process, enzyme medications are prescribed, the basis of which is pancreatin. The dose of medication is determined based on the amount of stool and the determination of neutral fat in the stool.
To combat the disease, the patient is prescribed mucolytics. These are special elements that soften bronchial mucus. Treatment must be continued throughout the patient's life. It consists not only in the use of medications, but also in carrying out physical procedures:

Bronchoscopy is a special event that allows you to effectively combat cystic fibrosis. The bronchial tree is washed using saline or mucolytics. If the patient has respiratory diseases, pneumonia or bronchial otitis, then antibacterial medications will be needed for treatment. The main symptom is digestive deficiency. Therefore, antibiotics are administered orally via aerosols or injection.
The main treatment option is lung transplantation. This is a serious operation; the question of carrying out the event arises when therapy has exhausted its capabilities. To improve the patient's quality of life, a double lung transplant will be needed.
This procedure will help if other organs in the body are not affected by the disease. Otherwise, serious intervention will not bring the expected effect.
Cystic fibrosis is considered a very complex disease. Symptoms and types of disease vary greatly. Cystic fibrosis is influenced by various factors, with age being the main one. The disease affects a person's life until his death. Progress has been made in the treatment of the disease, but the prognosis is still considered unfavorable. In more than half of cases with cystic fibrosis, death occurs. And also the life expectancy is short - from 20 to 40 years. In Western countries, with proper treatment, patients live up to 50 years on average.
Treatment of cystic fibrosis is a very difficult undertaking. The main task of doctors is to stop the development and progression of the disease. The treatment process is symptomatic only. Active prevention can prolong the patient’s life. To prevent cystic fibrosis from progressing, the following actions:

It is impossible for pathogenic bacteria to spread through the bronchi. They often contain thick mucus, so the bronchi need to be cleared of harmful accumulation. Treatment should occur not only during attacks, but also during the passive course of the disease. For chronic and acute processes they are used the following medications:

Women who suffer from cystic fibrosis find it extremely difficult to become pregnant. During pregnancy, the fetus may experience a number of complications; this poses a danger to the child and the mother herself. Now the disease cystic fibrosis is not fully understood, but there are many control methods that can extend the life of patients with a complex diagnosis.
A comprehensive genetic study that allows us to identify the 25 most common gene mutations in Russia CFTR leading to the development of a severe hereditary disease.
Research method
What biomaterial can be used for research?
Venous blood, buccal (cheek) epithelium.
How to properly prepare for research?
No special preparation is required.
General information about the study
Cystic fibrosis (synonym: cystic fibrosis) is one of the most common autosomal recessive hereditary human diseases. It is characterized by dysfunction of the epithelium of the respiratory tract, intestines, pancreas, sweat and gonads.
The cause of cystic fibrosis is mutations in the gene CFTR (cysticfibrosistransmembraneregulator) , encoding an ATP-binding protein that forms a channel for chloride ions in cell walls. Mutations lead to disruption of the transport of chlorine ions through the membranes of epithelial cells, which is accompanied by increased secretion of thick mucus and blockage of the excretory ducts of various glands.
There are several forms of cystic fibrosis:
Currently, in the Russian Federation, the diagnosis of cystic fibrosis is given to one out of 9,000 newborns (for comparison: in Europe, cystic fibrosis is diagnosed with a frequency of 1: 2,000 - 3,000 newborns). However, the form of mass screening of newborns adopted in Russia is imperfect and sometimes does not allow detection of the disease at the preclinical stage.
Every cell in our body has two copies of the gene. CFTR. One copy comes from the father, and the second from the mother. The disease cystic fibrosis is autosomal recessive, i.e. it develops only if the child receives a mutant gene from both father and mother CFTR. At the same time, parents who have second copies of the gene CFTR normal, do not suffer from cystic fibrosis and sometimes do not even realize that they are carriers of it. According to statistics, in the European population the carrier of gene mutations is CFTR on average every 25th person is.
There are approximately one thousand different mutations in the gene CFTR. They occur with varying frequencies in different populations. Some gene disorders may not have any manifestations. But most mutations cause a pathological effect, because they lead to disruption of the functioning of the protein.
This comprehensive study included gene analysis CFTR for 25 mutations, the most common in the Russian Federation, Eastern Europe and Scandinavia and associated with the development of severe clinical forms of cystic fibrosis. The study makes it possible to identify up to 95% of all possible patients, which significantly exceeds the resolution of screening approved in Russia.
The study will help not only confirm or refute the diagnosis of cystic fibrosis, but also identify carriers of the mutation in healthy people. It is especially important to conduct genetic testing in families in which there are patients with cystic fibrosis, since a couple where both parents are carriers of mutations has a 25% chance of having an affected child.
Until now, cystic fibrosis is considered an incurable disease, but early diagnosis and adequate therapy significantly improve the prognosis of the disease and prolong the patient’s life.
List of studied mutations in the gene CFTR:
When is the study scheduled?
What do the results mean?
The analysis examines 25 significant genetic markers of the gene CFTR, which makes it possible to detect the most common mutations leading to the development of the disease.
Based on the results of a comprehensive study, a conclusion from a geneticist is issued with an interpretation of the results.
Literature
The most common autosomal recessive childhood disorder among whites is cystic fibrosis or cystic fibrosis of the pancreas. The disease was first described in 1938 by American pathologist D. Anderson. According to foreign statistics, the frequency of cystic fibrosis in residents of Western Europe, on average, is 1 in 2-3 thousand newborns, in Russia it is less - 1 in 3-5 thousand. There are significant geographic and ethnic differences in the incidence of cystic fibrosis. In African and Asian countries, cystic fibrosis almost never occurs, while every 20th (5%) resident of Western Europe is a heterozygous carrier of a mutation in the cystic fibrosis gene ( CFTR). Such a high prevalence of mutant alleles in the gene CFTR associated with both the “founder effect” and the selective advantage of heterozygotes due to resistance to cholera, tuberculosis, and toxic forms of influenza. On the other hand, heterozygotes have twice the incidence of chronic pancreatitis.
The main pathogenetic mechanism of the disease is an increase in the viscosity of the secretion secreted by the mucus-forming glands of the bronchi, intestines, pancreas, and seminiferous tubules, accompanied by the closure of many ducts in these organs. In particular, the natural process of cleansing the bronchi is disrupted, which leads to their inflammation. Inflammation is accompanied by pulmonary edema and an increase in the production of abnormally viscous secretions. Increased viscosity of secretions in the gastrointestinal tract is accompanied by a change in the water-electrolyte component of pancreatic juice, its thickening and difficulty in releasing into the intestinal lumen. As a result, the formation of feces is disrupted, followed by intestinal obstruction and fibrocystic changes in pancreatic tissue occur.
The minimum diagnostic symptoms of cystic fibrosis are recurrent pulmonary, most often pseudomonas infections, dysfunction of the intestines and pancreas, and retardation in physical development. Characteristic signs of the disease are a large amount of indigestible fat in the patient’s coprogram and an increase in the concentration of sodium and chloride ions during a sweat test. Some patients already have intestinal obstruction at birth due to the presence of meconium ileus. Such patients require urgent surgical intervention. Sometimes meconium ileus in a fetus with cystic fibrosis can be detected by ultrasound examination already in the 2-3rd trimesters of pregnancy. There are three clinical forms of cystic fibrosis: pulmonary (15-20% of cases), intestinal (10%) and mixed. Most children suffering from cystic fibrosis in our country die in childhood, but erased forms of the disease that appear in adults have also been described.
The following groups of people are subject to examination for the presence of cystic fibrosis: (1) patients with recurrent bronchopulmonary diseases, Pseudomonas aeruginosa infection, asthma, and allergies; (2) persons with diseases of the gastrointestinal tract (prone to constipation, colitis, chronic pancreatitis, recurrent intestinal obstruction; newborns with meconium ileus, peritonitis; children with a large belly and low body weight (with normal appetite); patients with liver cirrhosis of unknown origin ; (3) men with infertility after excluding other causes (98% of men with cystic fibrosis have a narrowing of the vas deferens).
The cystic fibrosis gene was mapped in 1985 to the long arm of chromosome 7 at 7q31.2. In 1989, it was identified, and this discovery was accompanied by the simultaneous publication of three articles in one of the most prestigious journals in the world (Science). The first article was devoted to the cystic fibrosis gene itself. A group of Canadian researchers led by L. Ch. Tsui managed to isolate and clone the cDNA of the cystic fibrosis gene ( СFTR), its nucleotide sequence, exon-intron boundaries and regulatory regions of the gene were determined. The second article was devoted to the protein encoded by the gene СFTR and being the primary biochemical defect in patients with cystic fibrosis. It turned out that this is a protein that regulates the transmembrane conductivity of chlorine ions, localized on the apical membranes of the exocrine glands of the epithelium and performing the functions of a chlorine channel. The third article analyzed mutations in the gene СFTR. As soon as the nucleotide sequence of the coding region of a gene becomes known, one can immediately ask the question: what happened to the patient, how does the gene of a sick person differ from a normal gene? Using normal cDNA of the gene СFTR As a probe, it was possible to isolate and sequence mutant cDNA from the bronchial epithelium of one girl with cystic fibrosis. It turned out that this patient is homozygous for a specific mutation - a deletion of three nucleotides in the 10th exon of the gene, accompanied by the absence of phenylalanine at position 508 of the protein - delF508. The frequency of this mutation in patients with cystic fibrosis in Canada, North America and Northern Europe reaches 80%.
Currently, more than 1000 different mutations in the gene have been identified in patients with cystic fibrosis CFTR, mainly of the missense type. However, delF508 remains the most common. Its frequency in patients with cystic fibrosis in different populations varies from 30% to 80%. In Europe, there is a certain gradient in the spread of this mutation from north to south: in Denmark its frequency reaches 85%, in Italy it decreases to 50% and in Turkey to 20-30%. In Slavic populations, the frequency of delF508 among patients with cystic fibrosis is about 50%. Major mutations include missense mutations W1272X (found in more than 30% of cases in patients with cystic fibrosis belonging to the Ashkenazi Jewish ethnic group), G542X, G551D, R117H, R334W, etc. Thus, in 70-80% of cases, molecular diagnosis of mutations in gene CFTR turns out to be successful. This allows prenatal diagnosis of cystic fibrosis to be carried out already in the first trimester of pregnancy in order to prevent the rebirth of an affected child in a high-risk family.
In cases where it is not possible to carry out molecular identification of mutations in the gene CFTR in the patient or his heterozygous parents, prenatal diagnosis of cystic fibrosis can be carried out at 17-18 weeks of pregnancy by analyzing the activity in the amniotic fluid of a number of enzymes of intestinal origin - gammaglutamyl transpeptidase, aminopeptidase and the intestinal form of alkaline phosphatase. The presence of mucus plugs in the intestines of a fetus with cystic fibrosis at this stage leads to a decrease in the content of these enzymes in the amniotic fluid of a pregnant woman.
Intensive research is currently being conducted to identify connections between types of mutations in the gene CFTR and clinical polymorphism of the disease. Mutations associated with severe and mild forms of the disease have been identified. It was found that some mutations, including delF508, disrupt protein processing, as a result of which it does not reach the apical membrane and the chloride channel does not form. This explains the severe clinical picture of cystic fibrosis with such disorders. Other mutations (R117H, R334W, R347P), identified in milder forms of cystic fibrosis, do not affect protein processing; a chloride channel is formed, but works less intensively. For example, the missense mutation R117H was found in men suffering from infertility due to blockage of the seminiferous tubules. At the same time, the clinical picture of cystic fibrosis in such patients is usually absent or very blurred. That is, in carriers of the R117H mutation, the viscosity of the abnormal secretion secreted by the exocrine glands of the epithelium is increased so slightly that this does not lead to abnormal processes in the lungs, pancreas or intestines, but this increase is sufficient to form vas deferens obstruction.
Using the transgenosis technique, model lines of mice with mutations in the cystic fibrosis gene, including those that were identified in patients, were constructed in various laboratories in the USA and Great Britain. It has been shown that different mutations have different effects on the phenotype of animals. In mice of some transgenic lines, predominant damage to the lungs was noted, while in other lines - to the pancreas and intestines. In one line, the death of a large number of embryos was observed from causes similar to meconium ileus. Thus, these lines represent ideal models not only for studying the molecular basis of the pathogenesis of cystic fibrosis, but also for testing various therapeutic programs for this serious disease.
Currently, effective methods of etiopathogenetic treatment have been developed that can significantly increase the life expectancy of patients with cystic fibrosis. Treatment of patients with cystic fibrosis is indicated in specialized regional centers. The general treatment regimen includes the use of mucolytic agents, antimicrobial drugs, pancreatic enzymes, vitamins, and certainly the use of kinesiotherapy and physiotherapy. Management of patients with cystic fibrosis is carried out according to national recommendations .
Cystic fibrosis (cystic fibrosis) is a hereditary disease that is caused by a mutation in the cystic fibrosis transmembrane regulator gene. It manifests itself in systemic damage to the exocrine glands and is accompanied by severe dysfunction of the gastrointestinal tract, respiratory system and a number of other organs and systems.
| ICD-10 | E84 |
|---|---|
| ICD-9 | 277.0 |
| DiseasesDB | 3347 |
| MedlinePlus | 000107 |
| eMedicine | ped/535 |
| OMIM | 219700 |
| MeSH | D003550 |
The first mention of the disease dates back to 1905 - at that time, the Austrian doctor Karl Landsteiner, when describing cystic changes in the pancreas with meconium ileus in two children, expressed the idea of the relationship between these phenomena.
The disease was described in detail, identified as an independent nosological unit and proved its hereditary nature by the American pathologist Dorothy Anderson in 1938.
The name “cystic fibrosis” (from the Latin mucus - mucus, viscus - viscous) was proposed in 1946 by Sidney Farber, an American.
The incidence varies widely among different ethnic groups. Cystic fibrosis is most common in Europe (average 1:2000 - 1:2500), but the disease has been reported in representatives of all races. The incidence of cystic fibrosis in the indigenous population of Africa and Japan is 1:100,000. In Russia, the average prevalence of the disease is 1:10,000.
The gender of the child does not affect the incidence of the disease.
Inheritance occurs in an autosomal recessive manner. In carriers of one defective gene (allele), cystic fibrosis does not manifest itself. If both parents are carriers of the mutated gene, the risk of having a child with cystic fibrosis is 25%.
In Europe, every 30th person is a carrier of a defective gene.
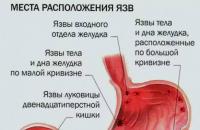
Depending on the location of the lesion, cystic fibrosis is divided into:
Separately, meconium ileus is distinguished, in which, as a result of reduced activity of pancreatic enzymes and insufficient production of the liquid part of the secretion by intestinal epithelial cells, meconium (original feces) adhered to the intestinal wall clogs the lumen and causes intestinal obstruction.
There are also types of mutations in the CFTR gene:
Cystic fibrosis is caused by mutations in the CFTR gene located on the long arm of chromosome 7. This gene is found in many animals (cows, mice, etc.). It contains about 250,000 base pairs and consists of 27 exons.
The protein encoded by this gene and responsible for the transport of chlorine and sodium ions across the cell membrane is located mainly in the epithelial cells of the respiratory tract, intestines, pancreas, salivary and sweat glands.
The CFTR gene itself was identified in 1989, and to date, about 2000 variants of its mutations and 200 polymorphisms (variable regions in the DNA sequence) have been discovered.
In representatives of the European race, the most common mutation is F508del. The maximum number of cases of this mutation was registered in the UK and Denmark (85%), and the minimum - among the population of the Middle East (up to 30%).
Some mutations are common in certain ethnic groups:
In Russia, 52% of mutations causing cystic fibrosis are the F508del mutation, 6.3% are the CFTRdele2.3(21kb) mutation, 2.7% are the W1282X mutation. There are also types of mutations such as N1303K, 2143delT, G542X, 2184insA, 3849+10kbC-T, R334W and S1196X, but their frequency does not exceed 2.4%.
The severity of the disease depends on the type of mutation, its localization in a particular region and the specificity of its effect on the function and structure of the encoded protein. The severe course of the disease and the presence of concomitant complications and exocrine pancreatic insufficiency are distinguished by mutations F508del, CFTRdele2,3(21kb), W1282X, N1303K and G542X.
Severe cases of cystic fibrosis also include the disease caused by the mutation DF508, G551D, R553X, 1677delTA, 621+1G-A and 1717-1G-A.
Cystic fibrosis caused by mutations R117H, 3849+10kbC-T, R 374P, T338I, G551S occurs in a milder form.
With the G85E, R334W and 5T mutations, the severity of the disease varies.
Mutations that block protein synthesis include mutations G542X, W1282X, R553X, 621+1G-T, 2143delT, 1677delTA.
Mutations that cause disruption of post-translational modification of proteins and their conversion into mature RNA (processing) include mutations DelF508, dI507, S549I, S549R, N1303K.
Mutations have also been identified:
As a result of the mutation, the structure and functions of the CFTR protein are disrupted, so the secretions of the endocrine glands (sweat, mucus, saliva) become thick and viscous. The content of protein and electrolytes in the secretion increases, the concentration of sodium, calcium and chlorine increases, and the evacuation of secretions from the excretory ducts becomes much more difficult.
As a result of retention of thick secretions, the ducts expand and small cysts form.
Constant stagnation of mucus (mucostasis) causes atrophy of glandular tissue and its gradual replacement with connective tissue (fibrosis), and early sclerotic changes in organs develop. With secondary infection, the disease is complicated by purulent inflammation.
Cystic fibrosis is caused by the inability of a defective protein to fully perform its functions.
As a result of protein dysfunction, an increased amount of chloride ions gradually accumulates in cells and the electrical potential of the cell changes.
A change in electrical potential causes sodium ions to enter the cell. An excess of sodium ions provokes increased absorption of water from the pericellular space, and a lack of water in the pericellular space causes thickening of the secretion of the exocrine glands.
When it is difficult to evacuate thick secretions, the bronchopulmonary and digestive systems are primarily affected.
Impaired patency of small bronchi and bronchioles leads to the development of chronic inflammation and destruction of the connective tissue framework. Further development of the disease is accompanied by the formation of saccular, cylindrical and “teardrop-shaped” bronchiectasis (dilatation of the bronchi) and emphysematous (inflated) areas of the lung.
Bronchiectasis is observed with equal frequency in the upper and lower lobes of the lungs. In most cases, they are not detected in children in the first month of life, but by the 6th month they are observed in 58% of cases, and after six months - in 100% of cases. At this age, various changes are found in the bronchi (catarrhal or diffuse bronchitis, endobronchitis).
The bronchial epithelium is exfoliated in some places, and foci of goblet cell hyperplasia and squamous metaplasia are observed.
When the bronchi are completely blocked by mucus, zones of collapse of the lung lobe (atelectasis) are formed, as well as sclerotic changes in the lung tissue (diffuse pneumosclerosis develops). In all layers of the bronchial wall there is infiltration of lymphocytes, neutrophils and plasma cells.
The mouths of the mucous bronchial glands dilate, purulent plugs are revealed in them, and in the lumens of bronchiectasis there is a large amount of fibrin, disintegrating leukocytes, necrotic bronchial epithelium and colonies of cocci. The muscle layer is atrophied, and the walls of bronchiectasis are thinned.
If a bacterial infection occurs against the background of impaired immunity, abscess formation begins and destructive changes develop (Pseudomonas aeruginosa is cultured in 30% of cases). With the accumulation of foam cells and eosinophilic masses with the inclusion of lipids, secondary lipoproteonosis develops due to disruption of homeostasis.
By age 24, pneumonia is detected in 82% of cases.
Life expectancy with cystic fibrosis depends on the state of the bronchopulmonary system, since the patient, due to progressive changes in the vessels of the pulmonary circulation, gradually decreases the amount of oxygen in the blood and the right parts of the heart increase and expand (the “pulmonary heart” develops).
Other changes in the heart area are also observed. Patients are diagnosed with:
Valvular and parietal endocarditis is possible.
When the secretion of the pancreas thickens, blockage of its ducts often occurs during the period of intrauterine development. In such cases, pancreatic enzymes produced by this gland in normal quantities are not able to reach the duodenum, so they accumulate and cause tissue breakdown in the gland itself. By the end of the first month of life, the pancreas of such patients is an accumulation of fibrous tissue and cysts.
The cyst occurs as a result of expansion of the interlobular and intralobular ducts and flattening and atrophy of the epithelium. Inside the lobules and between them, there is a proliferation of connective tissue and its infiltration by neutrophils and lymphohistiocytic elements. Hyperplasia of the islet apparatus, atrophy of the gland parenchyma and fatty degeneration of the tissue also develop.
The intestinal epithelium becomes flattened and includes an increased number of goblet cells, and mucus accumulation is present in the crypts. The mucous membrane is infiltrated with lymphoid cells including neutrophils.
Mutations that are accompanied by a decrease in the conductivity of chloride ions or the level of protein or normal RNA cause the slow development of chronic pancreatitis with relative preservation of pancreatic function for a long time.
Cystic fibrosis in newborns in 20% of cases leads to blockage of the distal parts of the small intestine with thick meconium.
In some cases, the disease is accompanied by prolonged neonatal jaundice, which is caused by the viscosity of bile and increased formation of bilirubin.
Almost all patients experience thickening of the connective tissue and scar changes in the liver (fibrosis). In 5-10% of cases, the pathology progresses and causes biliary cirrhosis and portal hypertension.
Also in the liver the presence of:
Cystic fibrosis is accompanied by an abnormal function of the sweat glands - the concentration of sodium and chlorine in the secretion is increased, and the amount of salt exceeds the norm by approximately 5 times. This pathology is observed throughout the patient’s life, so a hot climate is contraindicated for people suffering from cystic fibrosis (the risk of heat stroke increases, and convulsions are possible due to the development of metabolic alkalosis).
Cystic fibrosis in most cases manifests itself before one year of age.
In 10% of cases, symptoms of the disease (meconium ileus or meconium ileus) are detected by ultrasound examination during fetal development in the 2-3 trimester.
In some children, intestinal obstruction is detected in the first days of life. Signs of meconium ileus are:
Over the course of two days, the child’s condition worsens - pallor and dryness of the skin appear, tissue turgor decreases, lethargy and adynamia appear. Dehydration develops and intoxication increases. In some cases, complications may develop (intestinal perforation and peritonitis).
Intestinal cystic fibrosis manifests itself in most cases after the introduction of complementary foods or artificial feeding due to a deficiency of pancreatic enzymes. Symptoms of this form of the disease are:
Possible rectal prolapse when sitting on a potty (observed in 10-20% of patients).
There is often a feeling of dryness in the mouth, which occurs due to the viscosity of saliva, so eating dry food is difficult, and while eating, patients are forced to drink large quantities of liquid.
At the initial stages, appetite may be increased or normal, but due to digestive disorders, hypovitaminosis and malnutrition subsequently develop. As the disease develops, signs of cirrhosis and cholestatic hepatitis (increased fatigue, weight loss, jaundice, darkening of urine, disturbances in behavior and consciousness, abdominal pain, etc.) appear.
Cystic fibrosis of the lungs, due to overproduction of viscous secretions in the bronchopulmonary system, causes obstructive syndrome, which manifests itself:
An unproductive cough is possible.
The infectious-inflammatory process is chronic and recurrent. Complications are observed in the form of purulent-obstructive bronchitis and severe pneumonia with a tendency to abscess formation.
Symptoms of the pulmonary form of the disease are:
 with cystic fibrosis.
with cystic fibrosis. Bronchial contents usually include Pseudomonas aeruginosa, Staphylococcus aureus and Haemophilus influenzae. Flora may be resistant to antibiotics.
The pulmonary form is fatal due to severe respiratory and heart failure.
Signs of cystic fibrosis in the mixed form include symptoms of intestinal and pulmonary forms.
The erased forms of the disease are usually diagnosed in adulthood, since special types of mutations in the CFTR gene cause a milder course of the disease, and its symptoms coincide with those of sinusitis, recurrent bronchitis, chronic obstructive pulmonary disease, cirrhosis of the liver or male infertility.
Cystic fibrosis in adults often causes infertility. 97% of men with cystic fibrosis have congenital absence, atrophy, or obstruction of the spermatic cord, and most women with cystic fibrosis experience decreased fertility due to increased viscosity of cervical mucus. At the same time, some women retain their reproductive function. Mutations of the CFTR gene are also sometimes found in men who do not have signs of cystic fibrosis (the mutation in 80% of such cases results in aplasia of the vas deferens).
Cystic fibrosis does not affect mental development. The severity of the disease and its prognosis depend on the timing of the disease manifestation - the later the first symptoms appear, the milder the disease and the more favorable the prognosis.
Since cystic fibrosis, due to a large number of mutation variants, is characterized by polymorphism of clinical manifestations, the severity of the disease is assessed by the condition of the bronchopulmonary system. There are 4 stages:
The diagnosis of cystic fibrosis is based on:
Laboratory methods to detect cystic fibrosis in children include:
DNA diagnostics helps to most accurately diagnose cystic fibrosis. Typically used for research:
Possible use:
In most cases, the PCR (polymerase chain reaction) method is used for research. The most common types of mutations in the CFTR gene are detected using specially designed diagnostic kits that allow simultaneous detection of several mutations.
Instrumental examination methods also help to diagnose cystic fibrosis:
Cystic fibrosis is also diagnosed by examining duodenal contents, which helps identify a decrease in the amount of enzymes or their absence in duodenal juice.
Exocrine pancreatic function is assessed using a fecal pancreatic elastase 1 (E1) test. Cystic fibrosis is manifested by a significant decrease in the content of elastase 1 (a moderate decrease indicates the presence of chronic pancreatitis, pancreatic tumor, cholelithiasis or diabetes).
Cystic fibrosis can also be detected through prenatal diagnosis. DNA samples are isolated at 9-14 weeks of pregnancy from chorionic villus sampling. At later stages of family contact, amniotic fluid (16-21 weeks) or fetal blood obtained by cordocentesis (after 21 weeks) is used for diagnosis.
Prenatal diagnosis is carried out if both parents have mutations or if a sick child in the family is homozygous. Prenatal diagnosis is recommended even if mutations are present in only one parent. A similar mutation identified in the fetus requires differentiation between homozygous gene inactivation and asymptomatic heterozygous carriage. For differential diagnosis, at 17-18 weeks, a biochemical study of the amniotic fluid is carried out for the activity of aminopeptidase, gamma-glutamyl transpeptidase and the intestinal form of alkaline phosphatase (cystic fibrosis is characterized by a decrease in the amount of these intestinal enzymes).
If mutations of the CFTR gene cannot be identified, and a child with cystic fibrosis has already died, the fetus is examined using biochemical methods, since prenatal molecular genetic diagnosis is considered uninformative in this case.
It is preferable to treat cystic fibrosis in children in specialized centers, since patients need comprehensive medical care, including the assistance of doctors, kinesiotherapists and social workers.
Since cystic fibrosis as a genetic disease is incurable, the goal of therapy is to maintain a lifestyle that is as close as possible to the lifestyle of healthy children. Patients with cystic fibrosis need:
To treat malabsorption syndrome (loss of nutrients entering the digestive tract), caused by insufficiency of pancreatic enzymes, pancreatic enzymes are used in the form of microgranules (Creon 10000, Creon 25000). The drugs are taken with meals, and the dose is selected individually.
Since pancreatic insufficiency in cystic fibrosis is not completely corrected, the sufficiency of the dose is indicated by the normalization of stool character and frequency, as well as laboratory data (steatorrhea and creatorrhoea are not detected in the coprogram, the concentration of triglycerides is normalized in the lipid profile).
Respiratory cystic fibrosis requires the use of:
To treat Pseudomonas aeruginosa infection, antibiotics are usually given intravenously.
The criterion for discontinuation of antibiotic therapy is the return of the main symptoms of exacerbation to the initial state for the patient.
Cystic fibrosis is a contraindication to the use of antitussive drugs.
An effective treatment for progressive liver damage in cystic fibrosis has not currently been developed. Typically, patients with initial signs of liver damage are prescribed ursodeoxycholic acid at a dose of at least 15-30 mg/kg/day.
Since damage to lung tissue is affected by the body's excessive immune response, macrolides, nonsteroidal anti-inflammatory drugs, and systemic and local glucocorticoids are used as anti-inflammatory therapy.
Cystic fibrosis is a disease in which the patient needs regular detailed examinations, including examination of external respiratory function, coprogram, anthropometry, general urine and blood tests. Once a year, a chest x-ray, echocardiography and ultrasound of the abdominal organs are performed, bone age is determined, and immunological and biochemical blood tests are performed.
Found a mistake? Select it and click Ctrl + Enterprint version

Contents Dietary supplement based on an extract obtained from the Spanish beetle (or Spanish beetle...

Ekaterina MirimanovaSystem minus 60. RevolutionSystem minus 60 with Ekaterina Mirimanova"System minus 60. Revolution"...

Heaviness and bloating are the causes of both ordinary overeating and more serious problems with the digestive tract....

The second blood group, Rh-negative, appeared many years ago, when a person stopped being a hunter, and...

PSYCHOLOGICAL ASPECTS OF ANOREXIA PHENOMENON (EXPERIMENTAL STUDY) T. V. Tarasova, E. V. Arsentieva V...

Contents Since the skin in this area is thin, it is more prone to the appearance of various types of spots. Redness under...

vseslav Sat, 10/17/2015 - 20:50 Vasileostrovskaya station is one of the oldest stations...

Most often, the goat mushroom (Suillus bovinus) is called a goat mushroom in the common parlance of mushroom pickers. This tubular...

Every person has had dreams at least once in their life. Many do not attach any importance to them, but some...

Daedalus (crater). Diameter: 93 km Depth: 3 km (NASA photo) The moon has attracted the attention of people since ancient times. In II...

Presidents' Day is celebrated on the third Monday of February in the United States. Until the 70s, celebrations were dedicated to...

On the day of the last call, I want to say just one word to my beloved head teacher: “Thank you.” But how much is this...

Watermelon is considered a tasty and healthy berry; its seeds are especially useful. Small, dark brown or...

“Speech development in 6-year-old children” A child is not born with developed speech. It is impossible to definitively answer the question about...

Ekaterina MirimanovaSystem minus 60. RevolutionSystem minus 60 with Ekaterina Mirimanova“System minus 60....
Heaviness and bloating are the causes of both ordinary overeating and more serious digestive problems...