What is possible and not possible during the Nativity Fast?
In 2018, the Nativity Fast will begin on November 28. During this period, Orthodox believers prepare to celebrate Christmas...
Fanconi syndrome (or de Toni-Debreu-Fanconi) is a kidney disease that is characterized by impaired reabsorption of proteins and ions during the concentration of primary urine and associated metabolic disorders and constancy of the internal environment. Defects in the renal tubules cause glucose, phosphates, amino acids and bicarbonates to be excreted from the body.
Fanconi syndrome occurs in all parts of the world, and its occurrence rate is one in 350 thousand births. This is quite common if we take into account the planetary scale.
This disease can be either congenital or acquired as a result of the progression of other diseases. Scientists have not yet fully determined which genetic defect causes the appearance of such biochemical shifts. There is a theory that due to damage to the proteins included in the membranes of the renal tubules, their permeability is impaired. Another hypothesis states that the mutation affects enzymes that reabsorb glucose and trace minerals.
There are complete and incomplete Fanconi syndrome. If all three biochemical components are damaged, then they speak of a complete syndrome; if only two of the three, then this is an incomplete, or partial, syndrome.
Fanconi syndrome is rarely considered on its own. Most often, it is associated with diseases such as cystinosis, galactosemia, systemic or congenital glycogenosis, tyrosinemia and other storage pathologies. In addition, poisoning with phosphates, aminoglycosides, large doses of tetracyclines and heavy metals can induce the appearance of this nosology.
Amyloidosis and vitamin deficiency can also be precursors of the syndrome. And some authors are inclined to think that illness can be attributed to a severe degree of rickets-like pathologies.

Fanconi syndrome, or glucoaminophosphate diabetes, which is more common among domestic authors, develops due to impaired transport of substances through the membranes of the renal tubules. The cause of this condition has not yet been fully elucidated by pathophysiologists, but its consequences are well known.
Secondary changes in the body, reminiscent of rickets, are the result of prolonged acidosis and a decrease in the level of phosphorus in the blood, as well as a decrease in the amount of ATP (adenosine triphosphate).
There are congenital and acquired Fanconi syndrome. The first, as a rule, is combined with fermentopathy, fructose intolerance, and pathological accumulation of glycogen. Acquired syndrome is a consequence of taking toxic drugs, for example, chemotherapy, cytostatics or antiretroviral drugs. In addition, this symptom complex may appear after a kidney transplant, with amyloidosis and myeloma.

As a rule, primary or congenital, it is detected in childhood. In adults, Fanconi syndrome appears as a secondary disease after serious poisoning, difficult to tolerate treatment. It can also be a complication of systemic diseases.
With Fanconi syndrome, the patient excretes glucose, phosphorus, proteins and other necessary substances in the urine. In this case, the amount of fluid lost is greater than normal, and the density of urine decreases. Patients complain of weakness and pain in muscles and bones, lethargy, and drowsiness.
Most often, secondary Fanconi syndrome affects women after fifty-five years of age. Against the background of an already reduced calcium level, there is a loss of phosphorus and other trace elements. This provokes the development of osteoporosis and, as a consequence, compression fractures of the vertebrae and complex fractures of tubular bones. Ultimately, the patient may remain disabled.
Fanconi syndrome can be seen most often and clearly in children. Symptoms appear already in the first year of life and at first they are similar to those in adults:
Due to the constant loss of vitamins, microelements and sugars, the child begins to lag behind his peers in mental and physical development. The doctor may observe curvature of the leg bones, a decrease in muscle tone with subsequent atrophy, to the point that five-year-old children cannot walk independently. The disease progresses with age, and by puberty the child develops chronic renal failure.
Sometimes there are manifestations of the disease from other systems, for example, damage to the eyes, central nervous system, heart and blood vessels. There may be combined congenital anomalies of the genitourinary system, ENT organs and gastrointestinal tract.

Clinically, primary and secondary Fanconi syndrome are distinguished; accordingly, there are their types and subtypes.
Idiopathic syndrome is divided into:
The classification of secondary Fanconi syndrome is associated with the group of primary pathology.
Metabolic disorders:
Acquired kidney pathologies:
The syndrome is also observed in cases of intoxication with heavy metals, poisoning with organic poisons, antibiotics, toxic drugs, and third-fourth degree burns.
As can be seen from the above classification, the loss of essential microelements and proteins in the urine is rarely an independent disease; as a rule, it is a complication of other pathologies.

De Toni-Debreu-Fanconi symptoms in children and adults are a reason for further diagnostic search. In a biochemical blood test, the doctor can detect a decrease in calcium, phosphorus, sodium and other trace elements against the background of an increase in the level of alkaline phosphatase (ALP). Since bicarbonates are lost in the urine, oxidation of the internal environment is observed. In a general urine test, the presence of glucose, phosphorus, proteins and amino acids is noted. Urine has low density and there is a lot of it.
Of the instrumental diagnostic methods, bone radiography is necessarily used to identify osteoporosis and pathological deformations, as well as to examine foci of ossification in order to see the lag in bone age from the passport age.
In addition, in some patients it is recommended to use radioisotope studies or bone scintigraphy. Areas of active bone growth will appear darker on the screen. This will allow the doctor to determine the severity of the disease.
To determine the extent of damage to the kidney tissue, a biopsy is performed. It reveals the characteristic shape of the renal tubules in the form of a swan neck, atrophy of the tubular epithelium, and the presence of areas of tissue fibrosis.

What should a doctor do after a diagnosis of Fanconi syndrome? Treatment of children begins the sooner, the better. It is aimed at correcting electrolyte imbalance, replenishing the amount of proteins and buffer bases. Patients are recommended to drink plenty of fluids, a special diet high in microelements, limiting salt and introducing alkalizing foods into the diet, such as milk and fruit juices. In addition, children are recommended to eat more dried fruits.
In secondary Fanconi syndrome, symptoms disappear after treatment of the underlying disease. Sometimes, with severe bone deformities, consultation with trauma surgeons or orthopedists may be required. But this is only possible if there is a remission with the disappearance of all symptoms for more than one and a half years.
To prevent a primary disease before pregnancy, married couples with a compromised family history should undergo medical genetic counseling. The probability of having a sick child in a family if there is a tendency to this pathology is 25 percent.
De Toni-Debreu-Fanconi syndrome in children and adults ends disappointingly. Irreversible changes occur in the renal parenchyma, which lead to the development of chronic renal failure and the need for dialysis.

Unfortunately, the syndrome does not only occur in humans. Certain breeds of dogs may also exhibit Fanconi syndrome. Due to the fact that the breeding of purebred dogs is associated with the accumulation of genetic mutations, they are susceptible to various hereditary diseases. One of them may be Fanconi syndrome. In this case, the animal progressively loses weight and muscle atrophy is observed. Due to pain in the bones, the dog becomes lethargic, weak, stops running and playing, and tries to step on its paws less due to pain in the bones. Another symptom is excessive thirst and frequent urination.
Fanconi syndrome is a congenital metabolic pathology that involves the cessation of absorption (reabsorption) of glucose, phosphoric and carbonic acid salts, and amino acids by the renal tubules. As a result, a pathology is formed that can be classified as a special type of diabetes or rickets.
Another more detailed name for the disease - de Toni-Debreu-Fanconi syndrome or glucose-phosphate-amine diabetes - is associated with a clarification of the description of the disease by three authors. Since the first was the Swiss pediatrician Fanconi, who discovered characteristic signs of protein and glucose in the urine of a child with dwarfism and rickets, it is more popular in medicine to mention one name.
The disease often accompanies other hereditary metabolic pathologies (fructose intolerance, galactosemia, tyrosinemia). The prevalence is 1 case per 350 thousand live births.
Modern genetic studies have established the role of changes in chromosome 15 (encoded as 15q15.3). Inheritance of a mutant gene can occur in three types:
Mutations that occur for the first time are also found in children who do not have similar changes in their relatives.
Clinicians distinguish depending on the cause:
Secondary disease occurs:
The main pathology develops in the mitochondria of cells. These intracellular structures are the “energy production factory” for all activities. To obtain the necessary kilocalories, a phosphorylation process involving oxygen occurs in the mitochondria.
A biochemical reaction requires a set of enzymes for step-by-step transformations. But in Fanconi syndrome they are absent in the cells of the renal tubular epithelium. Consequently, mitochondria cannot supply the required amount of energy. Reabsorption of necessary substances into the blood suffers.

Electron microscopy view of mitochondria, insufficient energy production deprives the organ and tissue of the ability to function
Excreted in urine:
A deficiency of these substances is registered in the blood and the overall metabolism changes:
The child's skeleton shows signs of rickets. Closer to adulthood, a process of softening of bone tissue occurs - osteomalacia.
Symptoms of the disease are noticed in a child after 6 months of age:
A clearer picture is formed in the second year of life, and sometimes at 5-6 years. Here the first place is taken by:
The lag in physical and mental development is beyond doubt. The child grows up fearful and unsociable.

Up to a year, the child should be examined monthly by a pediatrician
Bone changes appear:
Bones become brittle. For this reason, the child often experiences fractures. The patient's height is shorter than his peers.
Based on the degree of metabolic disturbance and the severity of the patient’s condition, there are 2 clinical variants of Fanconi syndrome:
The result of the disease is:
In adulthood, a person develops secondary Fanconi syndrome. It manifests itself:
The disease is most severe in postmenopausal women, when hormonal influences on the electrolyte balance are added. Brittle bones lead to severe fractures:
This means complete disability, the impossibility of fusion of bone tissue. Kidney failure develops gradually. The glomerular epithelium atrophies and is replaced by scar tissue.

Compression fracture of the vertebrae is dangerous due to compression of the spinal cord
Detection of the disease is based on x-ray and biochemical laboratory methods. X-rays reveal:
Among the biochemical disorders detected in the blood:
Urine contains:
For an accurate diagnosis, it is necessary to distinguish the identified signs from rickets-like diseases and complications of hereditary and other diseases. They are distinguished by an additional more complete examination of blood, urine, kidney function, and bone marrow.
Such conditions include:

Differential diagnosis is carried out by a qualified specialist
Treatment consists of:
Doctors use:
To prevent irreversible pathology, the disease must be treated at the first manifestations. Therefore, you should carefully monitor the child’s development and unusual symptoms in an adult.
Fanconi syndrome is a rare congenital kidney dysfunction that leads to impaired reabsorption of nutrients (proteins, amino acids) during the accumulation of urine. This disrupts metabolism. This threatens various organ pathologies.
The disease can progress either independently or with other congenital anomalies. The risk group includes newborns and babies under one year of age. According to statistics, There is 1 sick newborn for every 350 thousand healthy people.. However, adults are also often affected by the disease, mainly due to other acquired pathologies.
This disease was first learned about in 1931 thanks to a Swiss doctor. Examining another patient with the characteristics of this disease, the man came to the conclusion that the symptoms should be classified as a separate classification. After analyzing, scientists confirmed his guesses and the syndrome was named after the doctor who discovered it.
To date, Fanconi syndrome has not been fully studied by experts and causes a lot of discussion and disagreement due to the small incidence rate. However, many agreed that the main reasons are:
There are two forms of the disease:
The first type refers to a disorder of the X chromosome, when the disease progresses at the gene level, which means that the first symptoms appear in infancy, namely:
If the symptoms are not eliminated in time, the disease will progress further and turn into a secondary form.

Regarding the secondary form, it is a consequence of other factors:
The doctor can suspect the development of the disease based on the patient’s complaints and in addition to the medical history, i.e. interviewing the patient and collecting data, the specialist must prescribe an examination that will confirm or refute the diagnosis.
Since Fanconi syndrome is primarily characterized by renal failure, the following are primarily prescribed:

Often this pathology can be confused with other diseases, for example, chronic pyelonephritis. And only changes in urine analysis and x-rays will give the correct diagnosis.
Since the pathology is chronic, it will not be possible to completely get rid of it. However, it is possible to suppress exacerbations for a while. For this, doctors recommend:
If not diagnosed and treated in a timely manner, the disease can lead to serious complications:
To avoid disastrous consequences, preventive measures must be taken for groups of people at risk. They are:
Prevention consists of regular examination of the kidneys, bones and cartilage tissue, as well as avoiding factors that provoke the disease.
Since animals are often susceptible to human diseases, Basenji dogs are no exception. This is historically associated with the artificial creation of the breed, due to which genetic mutations accumulate, thereby provoking diseases. Symptoms in animals are similar to humans:
In addition to the genetic factor as the main one, in recent years an acquired disease has been observed in animals after consuming treats produced in China. Since the exact cause has not yet been determined, there are only guesses:
The most susceptible are miniature dog breeds, weighing up to 10 kg. Fortunately, the animal is treatable and can often be saved.
Despite the danger this disease poses, scientists around the world are promoting the creation of a vaccine that stimulates kidney function. Today, there are many ways to help a patient improve his condition, put the disease into remission and prolong life.
Treatment: HCO 3 replacement and measures to combat renal failure.
Fanconi syndrome can be:
Hereditary Fanconi syndrome. This disease usually accompanies other genetic disorders, in particular cystinosis. Cystinosis is an inherited (autosomal recessive) metabolic disorder in which cystine accumulates in cells and tissues (and is not excreted excessively in the urine, as in cystinuria). In addition to renal tubular dysfunction, other complications of cystinosis develop: visual disturbances, hepatomegaly, hypothyroidism and other clinical manifestations.
Fanconi syndrome may also accompany Wilson's disease, galactosemia, glycogen storage disorders, oculocerebrorenal syndrome (Low's syndrome), mitochondrial cytopathies, and tyrosinemia. The mechanisms of inheritance vary depending on the disease with which Fanconi syndrome is associated.
Acquired Fanconi syndrome. This disorder can be caused by a variety of medications, including cancer chemotherapy, antiretroviral drugs, and older tetracycline. All of these drugs are nephrotoxic. Acquired Fanconi syndrome can also develop after kidney transplantation and in patients with multiple myeloma, amyloidosis or other chemical disorders, or vitamin D deficiency.
Various functional transport defects develop in the proximal tubules, including impaired reabsorption of glucose, phosphate, amino acids, HC03, uric acid, water, potassium and sodium. Generalized aminoaciduria is observed and, unlike cystinuria, increased excretion of cystine is its least manifestation. The underlying pathophysiological mechanism is unknown but is likely related to mitochondrial abnormalities. Low levels of phosphate in the blood cause rickets, which is exacerbated by decreased conversion of vitamin D to its active form in the proximal tubule.
In hereditary Fanconi syndrome, the main clinical manifestations - proximal tubular acidosis, hypophosphatemic rickets, hypokalemia, polyuria and polydipsia - usually develop in childhood.
When Fanconi syndrome develops in the setting of cystinosis, growth and development are often delayed. Focal depigmentation is detected in the retina. Interstitial nephritis develops, leading to progressive renal failure, which can lead to death before adolescence.
In the case of acquired Fanconi syndrome in adults, laboratory signs of renal tubular acidosis (proximal type 2), hypophosphatemia and hypokalemia are determined. They may present with symptoms of bone disease and muscle weakness.
The diagnosis is established by identifying pathology of renal function, especially glycosuria, phosphaturia or aminoaciduria. In cystinosis, slit-lamp examination may reveal cystine crystals in the cornea.
Apart from eliminating the effect of nephrotoxin, there is no specific treatment. Acidosis can be reduced with tablets or solutions of Na or KHCO 3 or citrates, for example, Schol's solution, which is prescribed at a dose of 1 mEq/kg 2-3 times a day or 5-15 ml after meals and at night. Decreased potassium levels may require potassium salt replacement therapy. Hypophosphatemic rickets can be cured. Kidney transplantation has shown to be effective in treating renal failure. However, when the underlying disease is cystinosis, progressive damage may extend to other organs and eventually lead to death.
Fanconi syndrome is a rare problem that is associated with metabolic disorders. Most often, the disorder is hereditary and is registered in infancy. Acquired pathology is diagnosed much less frequently and is associated with a good prognosis, unlike congenital pathology. The disease is accompanied by damage to the kidney tubules, the occurrence of rickets and acidosis. If left untreated, the disease is fatal.
The exact nature of the occurrence of congenital pathology is currently unknown. Presumably, the formation of the disorder is based on a mutation in the gene responsible for the performance of enzymes that regulate the metabolism of glucose and amino acids. A secondary problem arises against the background of other pathologies, including:
Symptoms of the lesion are associated with metabolic failure. Patients suffer from a lack of amino acids, phosphorus and bone destruction. The main manifestations of the disease depend both on the cause of its occurrence and on the degree of damage to internal organs. It is customary to distinguish between the clinical picture of a congenital problem, which is diagnosed in children, and a secondary one. It is caused by the excretion of trace elements, proteins and glucose in the urine.
Signs of Fanconi syndrome in sexually mature patients are usually associated with symptoms of the underlying disorder. Patients note:
Babies often suffer from congenital pathologies. Clinical signs are recorded already in the first year of life. The main manifestations of de Toni-Debreu-Fanconi syndrome in children include:

To confirm the presence of the disease, a comprehensive examination will be required. It includes general and biochemical blood and urine tests. A decrease in the concentration of calcium and phosphorus, as well as a change in pH value, is recorded. In urine there is an increase in the content of phosphates, calcium and glucose. X-rays can reveal changes in bone tissue. Differential diagnosis is carried out with ailments that can provoke rickets. These include juvenile nephronophthisis, cystinosis, diabetes mellitus and various poisonings.
Laboratory testing is highly informative. When conducting them, it is important to take into account the probable cause of the development of Fanconi syndrome. Urine tests are indicative, in which low molecular weight proteins are recorded. The appearance of these compounds in samples indicates damage to the glomerular apparatus and indirectly indicates the severity of the damage. In most cases, the presence of retinol-binding protein and beta-2-microglobulin is noted in urine. Comparison of the concentrations of these substances with the release of albumin by the kidneys makes it possible to presumably determine the exact location of nephron damage. Fanconi syndrome is also characterized by an increase in the ratio of protein to creatinine in the urine.
Blood tests show a significant increase in the level of alkaline phosphatase, an enzyme found in bones. The connection indicates increasing processes of osteomalacia. It is important to exclude the hepatic origin of the problem, since the substance is also found in hepatocytes. The value of ultrasound in the diagnosis of Fanconi syndrome is small, since it can only reveal the hyperechogenicity of the renal parenchyma, which is not specific.
Therapy is determined by both the cause of the disease and the intensity of its symptoms. In the case of the formation of Fanconi syndrome as a result of another pathological process, the fight against the disorder comes down to influencing the original source of the problem. In this case, an integrated approach is important. The congenital disease is difficult to treat. This is due to the lack of an accurate understanding of the nature of its origin. Treatment of the disease in children and adults comes down to the correction of electrolyte disturbances, as well as the elimination of acidosis, which provokes fatal complications. Combating the symptoms of Fanconi syndrome involves both conservative methods and more radical ones, which include surgery.
When correcting the patient’s condition, the following main goals are pursued:
To achieve the objectives, drugs of various groups are used. As a rule, when de Toni-Debreu-Fanconi syndrome is detected, hospitalization of the patient is required, since infusions are required. To correct the violations that have occurred, the following means are used:

Surgery is performed only when the patient’s condition is stable. It is justified in cases of significant bone deformations due to the development of rickets in children or osteomalacia in adults. In order for surgery to have the desired effect, it is necessary to stop the process of releasing phosphorus and calcium into the blood. That is why medicinal correction of the patient’s condition is so important. Surgical correction is carried out only if there is a stable remission lasting at least a year.
In some cases, the issue of kidney transplantation is acute, since in the process of removing metabolic products, the nephrons are significantly damaged. Transplantation is especially relevant for secondary Fanconi syndrome.
A properly selected diet is the basis for treating the disease, especially when it is detected in young patients. There are several recommendations to correct electrolyte disturbances and reduce the risks of fatal complications:
The disease is associated with a high mortality rate. In the early stages, it is possible to compensate for the occurring disorders, but the syndrome progresses, which is accompanied by severe electrolyte imbalance and renal failure. The prognosis when the disease is detected is unfavorable.
Preventive recommendations have not been developed to date, since the pathogenesis of the problem has not been studied. To prevent congenital anomalies, doctors advise contacting geneticists at the pregnancy planning stage. Adults need to prevent the development of diseases that can provoke the occurrence of the syndrome.

In 2018, the Nativity Fast will begin on November 28. During this period, Orthodox believers prepare to celebrate Christmas...

Starting a family is the dream of most women. They want to have a loving husband and a bunch of kids. But it's not always a relationship...

This article contains: the most powerful prayer for divorce - information taken from all over the world, the electronic network and...

Information site about icons, prayers, Orthodox traditions. Prayer for scandals and quarrels in the family, with husband, with children...

What would New Year be without champagne, tangerines, Olivier, aspic and everyone’s favorite “Herring under a fur coat”. With the last one...

Let's prepare the necessary ingredients for the cookies. The first thing to do is put the water to boil. We need...

Is it possible to register an employee for the position of financial director - chief accountant? The chief accountant claims...

The head of a small business can easily manage the budget independently. CHECKED! If you manage...

The creation of new projects involves a preliminary economic justification for their feasibility, subsequent...
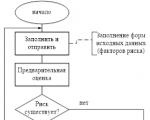
Reporting is generated by the RM, is agreed upon (approved) by the Risk Committee under the Management Board and transmitted to...

At the edge of a large, very large meadow, on a long emerald blade of grass lived a tiny Ladybug. God's little...

Nowadays, it is quite common for people to turn to the stars. With the help of a horoscope a person can find out...

Business or friendly. If you were pursuing a stranger, it means your level of trust in the female sex...

according to Freud's dream book If you dreamed about how you were fishing, it means that in real life you can hardly switch off...

Starting a family is the dream of most women. They want to have a loving husband and a bunch of kids. But it's not always...

This article contains: the most powerful prayer for divorce - information taken from all over the world, electronic...