What is possible and not possible during the Nativity Fast?
In 2018, the Nativity Fast will begin on November 28. During this period, Orthodox believers prepare to celebrate Christmas...
Currently, there are a huge number of hereditary diseases that a child receives along with genes from his mother or father. For some to manifest, it is necessary that both parents pass on the defective gene to their child. Thalassemia is one of these diseases. Few people know what kind of disease this is. In our article we will try to figure this out.
This is not even one, but a whole blood group that has recessive inheritance. That is, the child will receive it if both parents pass on the diseased gene to him. In this case, they say that there is homozygous thalassemia. The disease is characterized by disruption of the production of hemoglobin, which plays a major role in the transport of oxygen throughout the body.
Hemoglobin is a protein that has a protein part and a pigment part. The first consists of polypeptide chains: two alpha and two beta. Failure can occur in any of them, hence alpha thalassemia and beta thalassemia.
Impaired hemoglobin synthesis leads to a reduction in the lifespan of red blood cells, and this entails damage to cells and tissues. This process triggers a whole chain of reactions leading to the formation of various pathologies in the body.
There are several approaches to classifying this disease. If we consider in which circuit the failure occurred, we can distinguish:
In each case, the severity of symptoms may vary significantly. Taking this into account, we distinguish:
Depending on whether the child received the gene from both parents or from one, the disease is divided into:
All varieties are characterized by their symptoms and severity.
Each disease has its own causes; thalassemia is also formed under the influence of genes that the child receives from his parents. This genetic disease is particularly complex, but it is also the most common in the world.
Thalassemia is inherited in a recessive manner through the autosome of the parents. This means that the probability of getting sick is 100% for those who received defective genes for this trait from their mother and father.
The disease develops when a mutation occurs in the genes that are responsible for the synthesis of hemoglobin. The alpha form of this disease is quite common in the Mediterranean and Africa. Some people associate thalassemia with malaria, as these areas often experience outbreaks of the disease.
The blame is attributed to the fact that a mutation occurs in genes and thalassemia develops; the photo demonstrates that a large number of patients are found in Azerbaijan, approximately 10% of the total population. This confirms that the prevalence of the disease is associated with mutations, and climatic conditions also influence the mutation process.

If a child develops homozygous or thalassemia major, symptoms begin to appear almost immediately after birth. These include:

If a child is diagnosed with thalassemia, the symptoms are pronounced, then there is a high probability that he will not live to see his second birthday.
When the pathology is inherited from only one of the parents, we can talk about thalassemia minor or heterozygous. Since there is a second healthy gene in the genotype, it significantly smoothes out the manifestation of the disease, and symptoms may not appear at all or give a smoothed picture.
Thalassemia minor has the following main symptoms:
Despite the smoothed symptoms, the danger lies in the fact that the body’s susceptibility to all infections greatly increases.
Medicine has the opportunity to make a diagnosis of thalassemia at the early stages of development; diagnosis is carried out on the basis of laboratory blood tests. They immediately show that hemoglobin has a disturbed structure. You can even determine which of the chains there are deviations.
In young children, the signs of thalassemia appear quite clearly, so there is usually no difficulty in making such a diagnosis. Parents, before deciding to have a child, should undergo examination, especially if there is a carrier of the gene or a patient in the family.
It is possible to make a diagnosis of thalassemia already in the early stages of pregnancy; amniotic fluid is taken for analysis and examined. It will always contain fetal red blood cells, upon examination of which the presence of pathology can be determined.

Early diagnosis is very important because it is possible to begin treatment without waiting for the birth of the child, which will give the most effective result.
If the beta variety of the disease is formed, then the synthesis of hemoglobin beta chains is disrupted in the body. They are responsible for the production of hemoglobin A, which in an adult makes up 97% of the total number of molecules. If you look at beta thalassemia - what it is, then we can say, based on a blood test, that there is a decrease in the number of beta chains, but their quality does not suffer.
The reason is that they disrupt the functioning of the genes responsible for the synthesis of chains. It has now been established that there are not only mutations that cause disturbances in the functioning of genes, but there are also some sections of DNA that affect the manifestations of these mutations. As a result, it turns out that in people with the same mutations in the genes responsible for hemoglobin synthesis, the degree of manifestation of the disease can vary greatly.
The clinical picture of the disease can be different, depending on this, beta thalassemia is divided into several groups. Not everyone is familiar with the concept of thalassemia; that this disease depends on many genetic factors is also not known to everyone.
There are several gene states that control the production of beta chains:
Taking all this into account, the following types of thalassemia are distinguished:

In addition to beta, hemoglobin also contains alpha chains. If their synthesis is disrupted, then we can talk about a form such as alpha thalassemia. The disease is manifested by the formation of only the beta chain, and this risks the fact that hemoglobin of this structure will not be able to fulfill its main purpose - to carry oxygen.
The manifestations of the disease will depend on the severity of the mutation of the genes that control the synthesis of alpha chains. This process is usually under the control of two genes, one the child receives from the mother and the other from the father.
Depending on the degree of gene mutation, this form of the disease is divided into several groups:

If you have a mild form of alpha thalassemia, treatment may not be necessary, but with a severe form you will have to be under the supervision of doctors all your life. Only regular courses of therapy can improve a person’s quality of life.
We figured out what kind of disease thalassemia is. Now we need to focus on treatment. It can be noted that therapy is aimed at maintaining hemoglobin at the required level and eliminating the heavy load on the body from large amounts of iron. Treatment methods also include the following:

In addition to the listed treatment methods, symptomatic therapy is also carried out, which alleviates the patient’s condition.
For doctors and geneticists, if there is a diagnosis of thalassemia, it is clear that it is not curable. We have not yet found ways and methods to cope with this disease. But there are still measures to prevent it. The following preventive measures can be mentioned:
Every organism contains a huge number of mutating genes; it is practically impossible to predict where and when a mutation will appear. This is why genetic consultations exist to help married couples understand their ancestry, or rather, diseases that are passed on from one generation to another.
Depending on the severity and form of the disease, the prognosis may vary. Patients with thalassemia minor live a normal life, and its duration is practically no different from the life expectancy of healthy people.
With beta thalassemia, a small proportion of patients survive to adulthood.
The heterozygous form of the disease practically does not require treatment, but with the homozygous, and even severe form, it is necessary to undergo regular blood transfusions. Without this procedure, the patient’s life is almost impossible.
Unfortunately, thalassemia is currently one of those diseases that science has not yet learned to cope with. You can only keep it under control to some extent.
Medicine knows many diseases of a genetic nature that develop as a result of a peculiar “confluence” of circumstances. When both parents who carry certain abnormal genes pass on a disease to their child, it is called “genetic.” Thalassemia is such a disease.
Thalassemia is a genetic disease also called hemoglobinopathy. The disease is characterized by a violation of the synthesis of one of the components of hemoglobin - the protein part.
Hemoglobin (Hb) is the main component of red blood cells - erythrocytes. The main function of blood cells is to carry out the respiratory function of the body by transporting oxygen (O2) through the circulatory system. Also, red blood cells deliver “waste” carbon dioxide back to the lungs for removal from the body.
The structure of the hemoglobin molecule allows for the transport of gases, however, anomalies in its structure lead to disruption of this function.
Hemoglobin consists of two parts: the protein component and the pigment part. The polypeptide chains that form globin (the protein component) consist of two parts: two alpha polypeptide chains and two beta chains. Thus, we get four components, the coordinated work of which allows the red blood cell to perform its functions.
The genetic disease is caused by disturbances in these chains. Failures in α-components lead to the development of the disease alpha thalassemia. And disturbances in the β-chain provoke the occurrence of beta thalassemia.
In both types of disease, the process of hemoglobin synthesis is distorted, which leads to short-lived activity of red blood cells. As a result, the supply of oxygen to the cells and tissues of the body is disrupted. Oxygen starvation develops. The situation is extremely dangerous and provokes the occurrence of pathologies in human organs and tissues.
Depending on the location of the failure, several types of disease are distinguished:
The most common diagnosis is beta thalassemia. The type of disease is divided into homozygous and heterozygous. Based on the severity of symptoms, beta thalassemia is divided into three forms:

Homozygous thalassemia, or thalassemia major, develops if both the father and mother of the child were carriers of the defective gene. It is also called Cooley's anemia. By the first year of life, a child with a similar gene exhibits a number of symptoms.
Heterozygous thalassemia is caused by the presence of an unhealthy gene in only one of the parents. The disease is called “minor” because of its insignificant manifestation and ease of progression. It happens that the symptoms are so mild that they remain practically unnoticed throughout life.
Thalassemia intermedia is characterized by a non-classical course of homozygous thalassemia, in which obvious signs such as severe hemolytic anemia are absent. A person can do without blood transfusions.
The occurrence of beta thalassemia leads to the fact that “incorrect” beta globin predominates in the human blood, resulting in a decrease in the production of beta chains. The lack of these chains provokes the appearance of an excessive amount of alpha chains, which have a detrimental effect on the formation and further viability of red blood cells.
In the human bone marrow, most young blood cells die, and those that were able to survive and fully enter the bloodstream are at risk of destruction. Incomplete hemoglobin in such blood cells is weak and leads to the vulnerability of red blood cells. Also, such blood cells have a weakened membrane - a membrane that quickly collapses. This process is called hemolysis and leads to homolytic anemia.

Constant hypoxia of the body leads to developmental delays in the sick child.
Homozygous thalassemia is divided into three stages depending on the nature of the symptoms and the severity of the course:
Homozygous thalassemia results in the body's inability to produce hemoglobin alpha chains, resulting in fetal hemoglobin not being synthesized in the fetus. This form of the disease is incompatible with life and leads to fetal death or the development of dropsy, which leads to termination of pregnancy.
The genes that control the production of beta chains can be in different states:

Depending on the extent of the disease, the following types of beta thalassemia are distinguished:
Homozygous thalassemia is diagnosed either immediately after the birth of a child, or during the first 10-12 months of life. Symptoms of the disease rarely go unnoticed until the beginning of the second year.
However, in most cases, developmental disorders become widespread, which becomes the reason for diagnosing thalassemia.
Thalassemia is diagnosed if the following symptoms are present:


With the heterozygous type of thalassemia, it is not always possible to determine the presence of the disease. This is due to sluggish symptoms, usually of a general nature. The symptoms often resemble a number of other diseases, although patients often do not attach any importance to them. This type of disease can provoke symptoms similar to other diseases, however, the real cause is thalassemia, the diagnosis of which requires testing.
Symptoms characteristic of heterozygous thalassemia:

When diagnosing a disease, an important indicator is the presence of the disease in the patient’s family members.
Alpha thalassemia is diagnosed when the synthesis of hemoglobin alpha chains is impaired. An additional beta chain appears in their place. As a result, the main function of hemoglobin is disrupted: blood cells become unable to transport oxygen to cell tissue.
Symptoms depend on the degree of mutation of the genes that the child receives from his parents. Genes responsible for creating alpha chains can interfere with their formation to varying degrees.
Depending on the degree of gene mutation, alpha thalassemia disease is divided into forms:
The main criterion when determining the presence of thalassemia in a patient is the test results.

Diagnosis of thalassemia is based on the blood picture:
Treatment of thalassemia depends on its type and the nature of the disease.
Thalassemia major refers to excessive hematopoiesis in bone tissue. With this type of disease, the patient becomes completely dependent on blood transfusions.
Treatment is carried out in three stages. At the initial stage, transfusions are carried out at an accelerated pace: in 14-21 days the patient undergoes from eight to ten transfusions. The result of this treatment is an increase in hemoglobin to 120-140 g/l.
The next stage is to reduce the frequency of transfusions. The procedure is carried out every three weeks in the amount of 20 milliliters per kilogram of weight. The goal of this process remains to maintain hemoglobin levels within 100 g/l. Thus, the symptoms caused by the disease “Cooley's anemia” are suppressed.
Then the frequency of transfusions is determined depending on the patient's condition and the course of the disease. This procedure not only improves overall well-being, but also has a positive effect on the skeleton, the condition of the spleen and has a beneficial effect on the development of the child.
A serious disadvantage of this method of treatment is the accumulation of iron-containing pigment in the body. The situation leads to an enlarged liver, as well as a disruption in the absorption of glucose by cells.
To eliminate the problem, treatment includes the drug Desferal, which is administered intramuscularly in a certain amount. It depends on how much blood the patient is transfused, as well as the age of the child. Often the medicine is combined with small doses of ascorbic acid.

In most cases, the described methods lead to an improvement in the condition; however, recently, bone marrow transplantation to the patient has been widely practiced, which also has positive results.
For heterozygous thalassemia, emergency treatment is not required. Since most often the hemoglobin level is within the normal range, transfusions are not needed for such patients.
The spleen rarely increases to a critical size, so cases of organ removal are rare.
Desferal is used to treat elevated iron levels. The drug also helps prevent siderosis.
Siderosis is a disease characterized by sedimentation in the lungs as a result of elevated iron levels.
A decrease in hemoglobin levels is observed with immunity disorders, namely with its decrease. Pregnant women are also at risk. Folic acid is prescribed as treatment, the dosage of which is determined individually.
The use of iron-containing drugs is extremely dangerous for patients with this disease. The introduction of such drugs into the body can cause death in a patient who previously felt normal.
With thalassemia, doctors: a therapist and a hematologist are involved in diagnosing and prescribing treatment.
Often children can inherit from their parents not only characteristic traits and positive qualities, but also a hereditary disease.
The likelihood of getting any hereditary disease depends directly on the presence of defective genes in the parents. Thalassemia refers specifically to such genetic diseases that are inherited from parents to their children.
Thalassemia is a group of genetic blood diseases (ICD 10 code – D56), which causes an abnormal mutation of red blood cells, resulting in a disruption in the transport of oxygen molecules to the cells and tissues of the body.
Hemoglobin makes up the bulk of the blood cell and is the main substance for binding oxygen molecules and moving them throughout the body. With this disease, the entire process of synthesis of hemoglobin polypeptide chains is disrupted, which leads to defective and short-lived red blood cells.
As a result of such changes, there is a lack of oxygen in the tissues of the body, which leads to pathology and deterioration in the functionality of human organs.
The carriers of this disease are defective genes that are responsible for the construction of hemoglobin. Their inferiority lies in the impossibility of compiling the correct set of amino acids in the chromosomal chain.
Polypeptide chains are responsible for the performance of hemoglobin: alpha (α) and beta (β). Impaired synthesis in any of these chains can result in abnormal red blood cells.
There are two types of this disease based on the irregular abnormality of the polypeptide chains:
And also any type of thalassemia may differ in the way the mutant gene is acquired from the parents. Heterozygous thalassemia is acquired from only one parent and is usually a mild form of the disease. Homozygous thalassemia is acquired from both parents and can have a moderate or severe form of the disease.
Four genes (two genes from each parent) are responsible for building the alpha chain.
The severity of thalassemia disease will depend on the number of transmitted “sick” genes:
Only two genes (one from each parent) are involved in creating the beta chain in hemoglobin. When a mutation occurs in one or two genes of this chain, beta thalassemia appears.

There are three forms of this type of disease:
In forms of thalassemia minor and major, symptoms manifest differently. According to the method of acquisition, thalassemia minor is classified as heterozygous, that is, the thalassemia gene is transmitted from one parent. In this case, due to the healthy gene from the second parent, the entire overall picture of the disease will be slightly noticeable.
The main symptoms of the minor form include:
Moderate and severe forms of thalassemia are classified as homozygous, since defective genes are received from the father and mother at once.
Symptoms for these forms of the disease are pronounced:
Children with severe thalassemia rarely exceed the five-year mark.
During the transition to adolescence, patients experience additional symptoms:
Modern medicine is able to diagnose this blood disease in the early stages if the diagnosis is carried out correctly.
The first step is timely genetic consultation before planning pregnancy to determine the heredity of thalassemia in future parents. Prenatal diagnosis is possible as early as the eleventh week of pregnancy. Early detection of the disease will help to begin to fight thalassemia in advance.
Sometimes a family history and external examination of the patient allows one to suspect a blood disease in the early stages.
To confirm the diagnosis and begin treatment, the following tests are prescribed for thalassemia:
Thalassemia is still one of the rare treatable diseases. This is primarily due to the poor effectiveness and complexity of the treatment method.
With thalassemia minor, no special treatment is required, but there is an action plan to maintain the patient’s moderate condition:
For moderate and major forms of thalassemia, a number of treatment measures are being developed:

For patients with thalassemia, the prognosis depends on the severity of the disease:
The main goal of prevention is to identify the risk factor for inheriting thalassemia.
There is a set of measures aimed at preventing and preventing this disease:
Every person who does not know his family history is required to undergo a genetic test in advance to detect thalassemia in the blood. It is important to know that this disease is not acquired during life, the child is already born with thalassemia in his genes.
Accumulating in the skin and mucous membranes ( oral mucosa, conjunctiva of the eye), bilirubin gives them a characteristic yellowish tint. The presence of jaundice is typical for severe forms of the disease and is an unfavorable prognostic sign.
Enlarged spleen ( splenomegaly)
The spleen is the main organ responsible for removing old and damaged blood cells from the bloodstream. In thalassemia, the main number of red blood cells formed in the bone marrow and released into the systemic circulation are small in size and have a deformed surface. They cannot pass through the capillaries of the spleen, as a result of which they are retained and accumulate in it in large quantities, causing the organ to enlarge.
With prolonged splenomegaly, not only deformed red blood cells, but also other normal blood cells begin to be retained in the spleen ( platelets, leukocytes). They cannot pass through the capillaries of the organ, since they are completely filled and blocked by red blood cells. The result of this process is the development of hypersplenism, a pathological process characterized by the destruction of normal blood cells in the enlarged spleen.
Urate diathesis
One of the substances released into the bloodstream during the destruction of blood cells is purine. It is part of nucleic acids - DNA ( deoxyribonucleic acid) and RNA ( ribonucleic acid), which are part of the genetic apparatus of cells. Once purine enters the bloodstream, it is transported to the liver, where it is converted into uric acid.
With an increase in the concentration of uric acid and its salts ( urates) in the blood, they can form crystalline compounds that settle in various tissues of the body, causing their damage.
The accumulation of uric acid and its salts can manifest itself:
 The diagnosis and treatment of thalassemia is carried out by a hematologist, who, if necessary, can involve specialists from other fields of medicine.
The diagnosis and treatment of thalassemia is carried out by a hematologist, who, if necessary, can involve specialists from other fields of medicine. The main methods used in the process of diagnosing thalassemia are:
Blood collection procedure
Blood is taken in the morning, on an empty stomach. A nurse collects blood for analysis. To prevent infection of the skin of the fingertip ( most often the nameless one on the left hand) is treated with a cotton swab soaked in 70% alcohol, after which a special disposable game is used to make a puncture to a depth of 2 - 4 millimeters. The first drop is removed with a cotton swab, after which several milliliters of blood are drawn.
Blood test in the laboratory
Part of the collected blood is transferred to a glass slide and stained with a special dye, after which it is examined under a microscope. The number of red blood cells is determined ( and other blood cells), their shape, size, color.
Another option is to place the test material in a special device - a hematology analyzer, which automatically performs a quantitative count of all cellular elements of the blood. This method more accurately determines the number of blood cells, but does not provide information about their shape and structure.
Changes in complete blood count in thalassemia
| Index | What does it mean | Norm | Changes in thalassemia |
| Shape of red blood cells | The “extra” globin chains formed during thalassemia are not used in the synthesis of hemoglobin, but accumulate in the center of the cell, which, when examined under a light microscope, gives the red blood cell the characteristic appearance of the target. | The red blood cells are uniformly red, round, and all of the same size. | Red blood cells are oval or round in shape, representing light cells with a dark spot in the center ( target red blood cells). |
| Red blood cell size | As a result of disruption of hemoglobin formation, microcytosis develops, characterized by the formation of red blood cells of small diameter. | 7,5 – 8,3 µm. | 3 – 6 µm. |
| Average red blood cell volume ( MCV) | This indicator is calculated by a hematology analyzer by dividing the sum of the volumes of all cellular elements by the number of red blood cells. Provides more accurate information than simply determining the size of red blood cells when examined under a microscope. In children and elderly people, the average volume of red blood cells may be slightly increased, which is not a deviation from the norm. | 75 – 100 cubic micrometers ( µm 3). | Less than 70 µm 3 |
| Red blood cell count | In thalassemia, a large number of small red blood cells are produced, but most of them are very quickly destroyed in the spleen, resulting in a decrease in the total number of these cells in the blood. | Men(M): 4.0 – 5.0 x 10 12 /l. | Less than 4.0 x 10 12 /l. |
| Women
(AND
):
3.5 – 4.7 x 10 12 /l. | Less than 3.5 x 10 12 /l. | ||
| Total amount of hemoglobin | In thalassemia, hemoglobin synthesis is impaired to varying degrees ( depending on the form of the disease), as a result of which its total amount in the peripheral blood is reduced. | M: 130 – 170 g/l. | Depending on the form of the disease, it may be normal or reduced, up to 10 g/l. |
| AND: 120 – 150 g/l. | |||
| Average hemoglobin concentration in erythrocytes ( MCHC) | It is calculated by a hematology analyzer and provides more accurate data on the hemoglobin content in red blood cells, and not in the total volume of blood. Calculated by dividing total hemoglobin by the hematocrit. | 320 – 360 g/l. | Less than 300 g/l. |
| Platelet count | With a long course of the disease, the phenomenon of hypersplenism may develop, which is characterized by a decrease in the number of all blood cells, including platelets. | 180 – 320 x 10 9 /l. | Normal or reduced. |
| White blood cell count | With thalassemia, there is a tendency to frequent infectious diseases, which is characterized by an increase in the concentration of leukocytes. However, with the development of hypersplenism, their amount in the blood may decrease, so when assessing this laboratory indicator, the general condition of the patient should be taken into account. | 4.0 – 9.0 x 10 9 /l. | Varies depending on the severity of thalassemia and the general condition of the patient. |
| Reticulocyte count | Thalassemia is characterized by an increased process of hematopoiesis in the bone marrow, as a result of which a large number of young forms of red blood cells are released into the bloodstream. | M: 0,24 – 1,7%. | 2.5 – 4% or more. |
| AND: 0,12 – 2,05%. | 3 – 5% or more. | ||
| Hematocrit | This indicator reflects the ratio of the total volume of blood cellular elements to the volume of plasma. Since red blood cells are the main cellular elements of the blood, a decrease in their number and size in thalassemia will affect the hematocrit value. | M: 42 – 50%. | less than 32%. |
| AND: 38 – 47%. | less than 38%. | ||
| Color index | Displays the hemoglobin content in red blood cells. In thalassemia, the globin chains are concentrated in the center of the red blood cells and the amount of normal hemoglobin is reduced, resulting in a reduced color index. | 0,85 – 1,05. | 0.5 and below. |
| Erythrocyte sedimentation rate (ESR) | In the vascular bed, red blood cells are in a “suspended” state in the blood plasma. If you put blood in a test tube and add an anticoagulant ( substance that prevents blood clotting), then after some time the blood will divide into two layers - heavier red blood cells will settle to the bottom of the test tube, and lighter plasma will remain on the surface. ESR is determined by negative charges on the surface of red blood cell membranes, which repel each other, preventing cell sedimentation. With thalassemia, both the total number of red blood cells and their size are reduced, as a result of which the ESR will be significantly increased. | M: 3 – 10 mm/hour. | more than 10 mm/hour. |
| AND: 5 – 15 mm/hour. | more than 15 mm/hour. |
24 hours before blood collection it is necessary to exclude:
Before starting the procedure, the patient's arm is bandaged with a rubber band in the shoulder area ( the outflow of blood is disrupted, the veins of the arm become filled with blood and become more visible, which makes it easier to determine their location).
Having previously treated the site of the intended injection with a cotton swab soaked in alcohol, the nurse inserts a needle into the vein, to which an empty syringe is attached. The needle should be inserted towards the patient's body, which corresponds to the direction of blood flow in the vein ( this prevents blood clots from forming after the procedure).
While inserting a needle into a vein, the nurse constantly pulls back the syringe plunger. When the needle is in the vein ( as evidenced by the appearance of dark cherry-colored blood in the syringe), the tourniquet is removed from the patient’s shoulder and a few milliliters of blood are drawn into the syringe, after which a cotton ball with alcohol is pressed to the injection site and the needle is removed. The blood is transferred into a test tube and sent to the laboratory for analysis. The patient is asked to sit in the corridor for 15–20 minutes to avoid complications ( dizziness, loss of consciousness).
Biochemical parameters determined for thalassemia
| Index | What does it mean | Norm | Changes in thalassemia |
| Bilirubin level (total fraction) | The total amount of bilirubin in the blood is determined by adding the amount of unbound and bound forms. This indicator can be increased with increased breakdown of red blood cells or with diseases of the liver and biliary system, so its determination is indicative and should always be accompanied by the determination of individual bilirubin fractions. | 0.5 – 20.5 µmol/l. | Often elevated, but may be within normal limits. |
| Unconjugated bilirubin | This fraction of bilirubin increases with massive destruction of red blood cells in the spleen and in the vascular bed, as well as their precursors ( predominantly erythroblasts) in red bone marrow. | 4.5 – 17.1 µmol/l. | Always elevated, can reach several hundred micromoles in 1 liter of blood. |
| Serum iron level | As mentioned earlier, with thalassemia, the amount of iron in the blood increases due to increased absorption in the intestine, as well as as a result of transfusion of large volumes of donor blood. | M: 17.9 – 22.5 µmol/l. | Increased in severe forms of the disease, especially in combination with an enlarged spleen and liver. |
| AND: 14.3 – 17.9 µmol/l. | |||
| Alanine aminotransferase level (AlAT) and aspartate aminotransferase (ASAT) | These substances are found in liver cells ( hepatocytes) in large quantities. An increase in their level in the blood indicates the destruction of hepatocytes and the release of these enzymes into the blood. This may be due to the development of foci of hematopoiesis in the liver or the toxic effect of free iron. | M: up to 41 U/l. | With a long course of the disease, it can increase tens of times ( depending on the degree of liver tissue damage). |
| AND: up to 31 U/l. | |||
| Uric acid level | An increase in this indicator indicates an increased process of breakdown of blood cells in the spleen. | 2.5 – 8.3 mmol/l. | It can increase several times, especially with the development of hypersplenism. |
The following are used in the diagnosis of thalassemia:
The total iron-binding capacity of plasma reflects the number of free active transferrin centers in the blood. Blood is taken from a vein in compliance with all the rules described earlier. The essence of the method is quite simple - a predetermined excess amount of free iron is added to a test tube with blood. Some of the iron binds to the free active centers of transferrin, the rest is removed and its amount is determined using special instruments. Based on the data obtained, conclusions are drawn about the life insurance system.
Normal values of TLC are in the range from 45 to 77 µmol/l. With thalassemia, the amount of free iron in the blood is significantly higher than normal. All active centers of transferrin are in a bound state and are practically devoid of the ability to bind iron, as a result of which the TCI decreases.
Determination of serum ferritin concentration
Research in recent years has established that the amount of ferritin in plasma is directly dependent on the amount of free iron in the body - as it increases, the concentration of ferritin also increases.
Based on the described mechanism, many methods have been developed for determining the concentration of this protein in the blood. One of the most commonly used is radioimmunoassay. Blood for this study is taken from a vein, in the morning, on an empty stomach ( fence rules are described above).
The essence of the method is as follows - a special substance is fixed on a certain solid carrier ( antibody), which can selectively bind to ferritin. A sample of the blood being tested is added to it, and all the ferritin binds to this substance, forming a strong connection.
The next step is to add other specific antibodies to the solution, to which a radioactive label is attached ( the iodine atom is more commonly used). Free antibodies bind to ferritin and, when the solution is removed, are retained on the solid support.
The last step is a study in a special gamma counter, which allows you to determine the amount of radioactive iodine on the antibodies attached to ferritin. Based on the data obtained, conclusions are drawn about the concentration of ferritin in the blood.
The serum ferritin level depends on gender and is:
Determination of erythropoietin levels
The essence of the method is to determine the amount of erythropoietin in the blood plasma. Radioimmunoassay can also be used for this purpose. The technique and rules of implementation are the same, only instead of antibodies to ferritin, specific antibodies to erythropoietin are used.
The normal concentration of erythropoietin in the blood is 10 – 30 mIU/ml ( international milliunits in 1 milliliter). In thalassemia, this indicator is increased several times, which is due to excess production of erythropoietin by the kidneys.
Different organs and tissues absorb X-rays at different intensities, causing their shadow images to appear more or less clear on X-ray film. Bone tissue has the maximum absorption capacity in the human body, which is characterized by the lightest areas on x-rays. Air practically does not absorb X-rays and is defined as the darkest area in the image.
X-ray examination of patients with thalassemia can reveal:
Contraindications to performing an x-ray examination are:
The method is based on the ability of ultrasonic waves ( whose frequency exceeds 20,000 hertz) pass through body tissues and are partially reflected by them. The maximum degree of ultrasound reflection is determined at the boundary of two different media ( e.g. air and liquid, air and organ tissue, organ tissue and bone). In this way, it is possible to obtain an image of internal tissues and organs, to study their structure, density and consistency.
Modern ultrasound machines are quite compact and easy to use, so the examination can be carried out directly in the doctor’s office. The patient lies down on the couch and exposes the part of the body being examined. A special gel is applied to the surface of the skin, which fills microcracks in the skin ( the air they contain may interfere with testing), after which a sensor emitting ultrasound is applied to the skin.
The reflected ultrasound waves are recorded by a special receiver, and after computer processing, an image of the organ or area being examined appears on the screen.
When diagnosing thalassemia, the following is carried out:
The collection of material for research is carried out under sterile conditions using sterile instruments. Most often, bone marrow is taken from the sternum, but puncture of other flat bones is also possible ( pelvic bones, vertebrae).
After treating the puncture site with a solution of 70% alcohol, a syringe with a special needle is used to pierce the periosteum and the upper part of the bone. The needle is advanced 1–1.5 cm deep, after which 0.5–1 ml of bone marrow substance is collected. The needle is removed and the puncture site is covered with a sterile bandage. The resulting material is sent to the laboratory, where it is stained with special dyes and examined under a microscope.
When examining bone marrow puncture from patients with thalassemia, a pronounced increase in the number of cells, mainly erythrocyte precursors, is determined. They have a characteristic structure and size ( small light-colored cells with an accumulation of hemoglobin in the center).
The principle of the method is the formation of a large number of copies of a certain gene and its subsequent study. The study requires a small amount of biological material containing cells ( blood, saliva, urine, etc.). The material being studied is placed in a test tube, and a set of special enzymes and reagents is added to it, which activate the process of doubling a strictly defined gene on a specific chromosome, and only if this gene is present in the genetic apparatus of the cell.
In other words, in order to obtain copies of the gene encoding, for example, the formation of the globin a-chain, it is necessary that it be present on chromosome 16 and not be deformed. In this case, special substances recognize this gene, attach and copy it, after which the process is repeated many times. If this gene is not on the chromosome ( for a-thalassemia), no reactions occur.
The same principle is used to determine the presence or absence of all genes responsible for the formation of various globin chains. This helps to establish the form of thalassemia and determine the likelihood of transmitting the disease to descendants.
 Unfortunately, at the present stage of development of medicine, there is no medicine that can save a person from this disease. The hematopoietic stem cell transplant method shows great promise ( bone marrow transplants), however, its implementation is fraught with many difficulties and is not always possible. That is why the goal of treatment in most cases is to eliminate the symptoms of the disease and prevent the development of complications.
Unfortunately, at the present stage of development of medicine, there is no medicine that can save a person from this disease. The hematopoietic stem cell transplant method shows great promise ( bone marrow transplants), however, its implementation is fraught with many difficulties and is not always possible. That is why the goal of treatment in most cases is to eliminate the symptoms of the disease and prevent the development of complications. Mild forms of thalassemia often do not require treatment. Such patients are recommended to have a preventive general blood test every six months. In more severe forms, treatment should begin as early as possible, since a lack of oxygen in the body can lead to the development of irreversible changes in the internal organs.
The main directions in the treatment of thalassemia are:
The only effective way to increase the number of red blood cells and hemoglobin in the blood is donor blood transfusion. The target hemoglobin level is 100 – 120 g/l.
Indications for blood transfusion are:
Possible adverse reactions during blood transfusion are:
Methods to increase the level of red blood cells and hemoglobin
| Name of the technique | Mechanism of therapeutic action | Mode of application | Evaluation of treatment effectiveness |
| Whole blood transfusion | Donated blood contains all blood cells ( red blood cells, platelets, leukocytes and lymphocytes). The effects of donor blood transfusion are:
| Whole donor blood is prescribed quite rarely, due to many adverse reactions. The only reasonable indication is the development of hypersplenism with a decrease in the number of all blood cells. Blood transfusions are performed 1–3 times a month, depending on the form and severity of the disease. 500 or more milliliters of blood can be transfused at one time. |
|
| Red blood cell transfusion | The patient is given clean ( laundered) red blood cells. The therapeutic effects are the same as with whole blood transfusion, but this method is characterized by a significantly lower number of complications, which is due to a smaller number of foreign cells entering the body. | Depending on hemoglobin levels and the clinical picture of the disease, red blood cells are transfused from 1 to 3 – 4 times a month. The duration of treatment is lifelong. | The criteria for the effectiveness of treatment are the same as for transfusion of whole blood products. |
Drug treatment of excess iron in the body
| Drug name | Mechanism of therapeutic action | Directions for use and doses | Evaluation of treatment effectiveness |
| Desferal (Deferoxamine) | This drug has the ability to bind both free iron and iron contained in ferritin and transferrin. The complexes formed in this case are non-toxic and are quickly eliminated from the body. | Prescribed when serum ferritin increases above 1000 mcg/l. The drug is administered intravenously, by drip, at a dose of 20–40 mg/kg/day, at least 5 days a week. The duration of treatment is lifelong. | Treatment effectiveness criteria are:
|
| Exjad | The mechanism of action is the same as that of Desferal, however, this drug fixes iron more selectively, with virtually no effect on the exchange of other microelements in the blood. | Take orally, 1 time per day ( preferably at the same time), 30 minutes before meals. The initial dose is 10 mg/kg, if necessary, can be increased to 30 mg/kg. The duration of treatment is at least 1 year. | |
| Vitamin C | Promotes the removal of iron from the body. | Take orally, 2 – 3 times a day. The recommended dose is 1 – 3 mg/kg/day. It is not recommended to prescribe simultaneously with Desferal. | The criteria for the effectiveness of treatment are the same as when taking Desferal. |
Drug treatment of urate diathesis
| Name of the medication | Mechanism of therapeutic action | Directions for use and doses | Evaluation of treatment effectiveness |
| Allopurinol | An antigout drug used to reduce the amount of urate in the blood. The main effects are:
| Inside, 3 times a day, after meals. The initial dose is 70 – 130 mg. If necessary, the dose can be increased to 200 mg. The treatment is long-term. | Treatment effectiveness criteria are:
|
| Benemid | A uricosuric drug that increases the excretion of uric acid in the urine. In addition, it increases the amount of urine excreted per day, which prevents the formation of stones in the kidneys and bladder. | Inside, after eating. The initial dose is 0.5 g/day. The dose is gradually increased until the concentration of uric acid in the blood normalizes ( maximum 3 g/day). Lifelong treatment. | Treatment effectiveness criteria are:
|
Indications for surgical removal of the spleen for thalassemia are:
The operation is performed under general anesthesia. The access is most often laparotomic - an incision is made in the abdomen, the vessels of the spleen are first ligated and then cut, and the organ is removed. The incision site is sutured with sterile threads and a sterile bandage is applied, which is changed every day. After surgery, it is recommended to avoid physical activity for at least one month.
Carrying out a bone marrow transplant includes many stages, the main ones being:
Thalassemia occurs when there is a mutation in the genes that are responsible for the synthesis of peptides - the main elements of hemoglobin. The severity of the disease depends on the number of mutated genes. Thalassemia leads to a deficiency of hemoglobin, a protein that plays a key role in transporting oxygen and carbon dioxide. Hemoglobin molecules contain two alpha globin and beta globin. The gene mutation leads to changes in blood composition and subsequent death of red blood cells.
Thalassemia causes the following effects in the body:
Thalassemia is a hereditary disease, meaning it is passed on to the child from the parents. The risk of developing the disease increases if it has previously been observed in one of your close relatives. Ethnicity is also a risk factor. Thalassemia is most common in the countries of the Mediterranean, Central Asia and Africa.
There are two main types of thalassemia: alpha and beta. The first of them involves a mutation of the genes responsible for the synthesis of alpha globins, and the second - of beta globins. The severity of the disease and its main features will depend on the type of genes affected, as well as on the number of pathological sections of DNA. Considering that alpha globins are encoded by four different genes, the following types of the disease are distinguished:
As for beta globins, they are encoded by a single gene located on chromosome 11. If there is a defective gene in only one of the paired chromosomes, a mild degree of the disease develops - thalassemia minor. If both chromosomes are affected, a serious disease occurs - thalassemia major (Cooley's disease).
Modern medical technologies make it possible to detect thalassemia during prenatal diagnosis. In this case, the expectant mother may be prescribed special treatment that can eliminate the pathology before the birth of the child. In born children, signs of thalassemia appear depending on the type of disease and the number of mutations. In some cases, symptoms are recorded immediately after birth, and in others - during the first two years of life.
The following are common symptoms of the disease:
Most often, symptoms characteristic of thalassemia do not allow a confident diagnosis. To do this, you need to undergo a special examination. First of all, the patient undergoes a general blood test, which reveals a decrease in the number of red blood cells and the appearance of smaller cells of different sizes and shapes. In addition to mature cells, their precursors, blasts, may be present in the blood.
Other specific blood tests are also prescribed to determine the severity of the disease. Special molecular tests (PCR) may also be prescribed, which help identify mutations at the gene and cellular level. In addition to blood tests, diagnostics of the liver and spleen are required - the organs most affected by thalassemia. In addition, it is necessary to check the condition of the bone tissue. For this purpose, ultrasound and radiography are performed.
It is extremely important to try to diagnose thalassemia in a child during pregnancy, especially if one or both parents have previously had this disease or are potential carriers of it. In this case, diagnostic methods are used such as chorionic villus biopsy (at the 11th week of pregnancy) and amniotic fluid sampling - amniocentesis (prescribed at the 16th week).
When a diagnosis such as thalassemia is made, therapy is prescribed depending on the stage and severity of the disease. If the symptoms are mild or absent altogether, no treatment is carried out. The patient only needs periodic blood transfusions, especially after surgery and childbirth, as well as to prevent various complications. At the same time, people with beta thalassemia require more frequent blood transfusions, and in order to normalize the increased level of iron in the body, they are forced to take specific drugs that bind and remove this substance.
If the disease occurs in a pronounced or severe form, the following treatment methods are used:
The mild form of thalassemia has a fairly good prognosis. These patients do not require ongoing treatment and complications are extremely rare. Moderate and severe forms of the disease also have a favorable prognosis, but for this the patient must undergo regular blood transfusions and use iron-binding drugs.
In most cases, death occurs due to excessive accumulation of iron in the body, as well as disruption of vital organs. However, even negative consequences can be dealt with through surgery. As practice shows, most people who follow the doctor's instructions live a completely normal life.
If a diagnosis such as thalassemia has been made, it is very important to take a responsible approach to starting a family. In case of pregnancy, the expectant mother must undergo a blood test and ultrasound according to a special schedule in order to ensure as early as possible the presence or absence of disease in the fetus. Otherwise, it is recommended to avoid taking any vitamin-mineral complexes or dietary supplements that contain iron. Patients are also prescribed a balanced and varied diet. Finally, they are obliged to carry out timely prevention of infectious diseases through vaccination.

In 2018, the Nativity Fast will begin on November 28. During this period, Orthodox believers prepare to celebrate Christmas...

Starting a family is the dream of most women. They want to have a loving husband and a bunch of kids. But it's not always a relationship...

This article contains: the most powerful prayer for divorce - information taken from all over the world, the electronic network and...

Information site about icons, prayers, Orthodox traditions. Prayer for scandals and quarrels in the family, with husband, with children...

What would New Year be without champagne, tangerines, Olivier, aspic and everyone’s favorite “Herring under a fur coat”. With the last one...

Let's prepare the necessary ingredients for the cookies. The first thing to do is put the water to boil. We need...

Is it possible to register an employee for the position of financial director - chief accountant? The chief accountant claims...

The head of a small business can easily manage the budget independently. CHECKED! If you manage...

The creation of new projects involves a preliminary economic justification for their feasibility, subsequent...
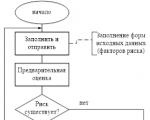
Reporting is generated by the RM, is agreed upon (approved) by the Risk Committee under the Management Board and transmitted to...

At the edge of a large, very large meadow, on a long emerald blade of grass lived a tiny Ladybug. God's little...

Nowadays, it is quite common for people to turn to the stars. With the help of a horoscope a person can find out...

Business or friendly. If you were pursuing a stranger, it means your level of trust in the female sex...

according to Freud's dream book If you dreamed about how you were fishing, it means that in real life you can hardly switch off...

Starting a family is the dream of most women. They want to have a loving husband and a bunch of kids. But it's not always...

This article contains: the most powerful prayer for divorce - information taken from all over the world, electronic...