What is possible and not possible during the Nativity Fast?
In 2018, the Nativity Fast will begin on November 28. During this period, Orthodox believers prepare to celebrate Christmas...
The process of creating new medicines is carried out in accordance with the international standards of GLP (Good Laboratory Practice), GMP (Good Manufacturing Practice) and GCP (Good Clinical Practice).
Clinical drug trials involve the systematic study of an investigational drug in humans to test its therapeutic effect or detect an adverse reaction, and the study of absorption, distribution, metabolism and excretion from the body to determine its effectiveness and safety.
Clinical trials of a drug are a necessary stage in the development of any new drug, or the expansion of indications for the use of a drug already known to doctors. At the initial stages of drug development, chemical, physical, biological, microbiological, pharmacological, toxicological and other studies are carried out on tissues (in vitro) or on laboratory animals. These are so-called preclinical studies, the purpose of which is to obtain scientific estimates and evidence of the effectiveness and safety of drugs. However, these studies cannot provide reliable information about how the drugs being studied will act in humans, since the organism of laboratory animals differs from humans both in pharmacokinetic characteristics and in the response of organs and systems to drugs. Therefore, clinical trials of drugs in humans are necessary.
A clinical study (test) of a medicinal product is a systemic study of a medicinal product through its use in humans (patient or healthy volunteer) in order to assess its safety and effectiveness, as well as to identify or confirm its clinical, pharmacological, pharmacodynamic properties, assess absorption, distribution, metabolism, excretion and interaction with other drugs. The decision to initiate a clinical trial is made by the customer, who is responsible for organizing, monitoring and financing the trial. Responsibility for the practical conduct of the research rests with the researcher. As a rule, the sponsor is a pharmaceutical company that develops drugs, but a researcher can also act as a sponsor if the study was initiated on his initiative and he bears full responsibility for its conduct.
Clinical trials must be conducted in accordance with the fundamental ethical principles of the Declaration of Helsinki, GСP (Good Clinical Practice) and applicable regulatory requirements. Before the start of a clinical trial, an assessment must be made of the relationship between the foreseeable risk and the expected benefit for the subject and society. The principle of priority of the rights, safety and health of the subject over the interests of science and society is put at the forefront. The subject can be included in the study only on the basis of voluntary informed consent (IS), obtained after a detailed review of the study materials. Patients (volunteers) participating in testing a new drug must receive information about the essence and possible consequences of the tests, the expected effectiveness of the drug, the degree of risk, enter into a life and health insurance agreement in the manner prescribed by law, and during the tests be under constant supervision of qualified personnel. In the event of a threat to the health or life of the patient, as well as at the request of the patient or his legal representative, the head of the clinical trial is obliged to suspend the trial. In addition, clinical trials are suspended if a drug is unavailable or insufficiently effective, or if ethical standards are violated.
The first stage of clinical trials of the drug is carried out on 30 - 50 volunteers. The next stage is expanded trials on the basis of 2 - 5 clinics involving a large number (several thousand) of patients. At the same time, individual patient cards are filled out with a detailed description of the results of various studies - blood tests, urine tests, ultrasound, etc.
Each drug undergoes 4 phases (stages) of clinical trials.
Phase I. First experience of using a new active substance in humans. Most often, studies begin with volunteers (healthy adult men). The main goal of the research is to decide whether to continue working on a new drug and, if possible, to establish the doses that will be used in patients during phase II clinical trials. During this phase, researchers obtain preliminary data on the safety of the new drug and describe its pharmacokinetics and pharmacodynamics in humans for the first time. Sometimes it is impossible to conduct phase I studies in healthy volunteers due to the toxicity of this drug (treatment of cancer, AIDS). In this case, non-therapeutic studies are carried out with the participation of patients with this pathology in specialized institutions.
Phase II. This is usually the first experience of use in patients with the disease for which the drug is intended to be used. The second phase is divided into IIa and IIb. Phase IIa are therapeutic pilot studies because the results obtained from them provide optimal planning for subsequent studies. Phase IIb studies are larger studies in patients with the disease that is the primary indication for the new drug. The main goal is to prove the effectiveness and safety of the drug. The results of these studies (pivotal trial) serve as the basis for planning phase III studies.
Phase III. Multicentre trials involving large (and, if possible, diverse) patient groups (average 1000-3000 people). The main goal is to obtain additional data on the safety and effectiveness of various forms of the drug, the nature of the most common adverse reactions, etc. Most often, clinical studies of this phase are double-blind, controlled, randomized, and the research conditions are as close as possible to normal real routine medical practice. Data obtained in phase III clinical trials are the basis for creating instructions for the use of the drug and for deciding on its registration by the Pharmacological Committee. A recommendation for clinical use in medical practice is considered justified if the new drug:
Phase IV. Studies are conducted after the drug is marketed in order to obtain more detailed information about long-term use in various patient groups and with various risk factors, etc. and thus more fully evaluate the drug strategy. The study involves a large number of patients, which makes it possible to identify previously unknown and rare adverse events.
If a drug is going to be used for a new indication that has not yet been registered, then additional studies are conducted, starting with phase II. Most often in practice, an open study is carried out, in which the doctor and the patient know the method of treatment (the study drug or a comparison drug).
When testing with a single-blind method, the patient does not know which drug he is taking (it may be a placebo), and when using a double-blind method, neither the patient nor the doctor is aware of this, but only the leader of the trial (in a modern clinical trial of a new drug, four parties: the sponsor of the study (most often this is a pharmaceutical manufacturing company), the monitor - a contract research organization, a doctor-researcher, a patient). In addition, triple-blind studies are possible, when neither the doctor, nor the patient, nor those who organize the study and process its data know the assigned treatment for a particular patient.
If doctors know which patient is being treated with which drug, they may spontaneously evaluate treatment based on their preferences or explanations. The use of blind methods increases the reliability of the results of a clinical trial, eliminating the influence of subjective factors. If the patient knows that he is receiving a promising new drug, the effect of treatment may be associated with his reassurance, satisfaction that the most desired treatment possible has been achieved.
Placebo (Latin placere - to like, to appreciate) means a drug that obviously does not have any healing properties. The Large Encyclopedic Dictionary defines placebo as “a dosage form containing neutral substances. Used to study the role of suggestion in the therapeutic effect of any medicinal substance, as a control when studying the effectiveness of new drugs.” test quality medicine drug
Negative placebo effects are called nocebo. If the patient knows what side effects the drug has, then in 77% of cases they occur when he takes a placebo. Belief in a particular effect can cause side effects to occur. According to the commentary of the World Medical Association to Article 29 of the Declaration of Helsinki, “...the use of placebo is justified if it does not lead to an increased risk of causing serious or irreversible harm to health...”, that is, if the patient is not left without effective treatment.
There is a term for “completely blinded studies” when all parties to the study are blinded to the type of treatment being given to a particular patient until the results are analyzed.
Randomized controlled trials serve as the standard of quality for scientific research into the effectiveness of treatments. The study first selects patients from a large population of people with the condition being studied. These patients are then randomly divided into two groups matched according to the main prognostic features. Groups are formed randomly (randomization) using tables of random numbers in which each digit or each combination of digits has an equal probability of selection. This means that patients in one group will, on average, have the same characteristics as patients in another. In addition, before randomization, it should be ensured that disease characteristics known to have a strong influence on outcome occur at equal frequencies in the treatment and control groups. To do this, you must first distribute patients into subgroups with the same prognosis and only then randomize them separately in each subgroup - stratified randomization. The experimental group (treatment group) receives an intervention that is expected to be beneficial. The control group (comparison group) is in exactly the same conditions as the first group, except that its patients are not exposed to the intervention being studied.
GOST R 56701-2015
NATIONAL STANDARD OF THE RUSSIAN FEDERATION
MEDICINES FOR MEDICAL USE
Guidance on planning preclinical safety studies for subsequent clinical trials and drug registration
Medicines for medical applications. Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals
OKS 11.020
11.120.01
Date of introduction 2016-07-01
Preface
1 PREPARED by the Technical Committee for Standardization TC 458 “Development, production and quality control of medicinal products” based on its own authentic translation into Russian of the document specified in paragraph 4
2 INTRODUCED by the Technical Committee for Standardization TC 458 “Development, production and quality control of medicines”
3 APPROVED AND ENTERED INTO EFFECT by Order of the Federal Agency for Technical Regulation and Metrology dated November 11, 2015 N 1762-st.
4 This standard is identical to the international document ICH M3(R2):2009* “Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals"). The name of this standard has been changed relative to the name of the specified international document to align with the names adopted in the existing set of standards “Medicines for medical use”. When applying this standard, it is recommended to use instead of reference international standards the corresponding national standards of the Russian Federation specified in Appendix DA
________________
* Access to international and foreign documents mentioned in the text can be obtained by contacting Customer Support. - Database manufacturer's note.
5 INTRODUCED FOR THE FIRST TIME
The rules for the application of this standard are established in GOST R 1.0-2012 (section 8). Information about changes to this standard is published in the annual (as of January 1 of the current year) information index "National Standards", and the official text of changes and amendments is published in the monthly information index "National Standards". In case of revision (replacement) or cancellation of this standard, the corresponding notice will be published in the next issue of the monthly information index "National Standards". Relevant information, notices and texts are also posted in the public information system - on the official website of the Federal Agency for Technical Regulation and Metrology on the Internet (www.gost.ru)
Introduction
The purpose of this standard is to establish common approaches to planning preclinical studies of medicinal products with the countries of the European Union, the United States of America, Japan and other countries that apply international ICH guidelines to justify the possibility of conducting clinical studies of a certain nature and duration, as well as subsequent state registration.
The standard promotes the timely conduct of clinical trials, reducing the use of laboratory animals in accordance with the 3R principle (reduce/refine/replace), and reducing the use of other resources in drug development. New alternative methods should be considered in vitro for safety assessment. These methods, if properly validated and accepted by all regulatory authorities in countries implementing ICH guidelines, can be used to replace existing standard methods.
This standard promotes the safe, ethical development of medicines and their availability to patients.
Preclinical safety assessment carried out for the purpose of state registration of drugs usually includes the following stages: pharmacological studies, general toxicological studies, toxicokinetic and preclinical pharmacokinetic studies, reproductive toxicity studies, genotoxicity studies. For drugs that have certain properties or are intended for long-term use, an assessment of carcinogenic potential is also necessary. The need for other preclinical studies to assess phototoxicity, immunotoxicity, toxicity in immature animals and the occurrence of drug dependence is determined on an individual basis. This standard specifies the need for nonclinical studies and their relationship to subsequent clinical studies in humans.
To date, countries using ICH guidelines have made significant progress in harmonizing the timing of nonclinical safety studies for clinical trials of medicinal products described in this standard. However, in some areas differences remain. Regulators and manufacturers continue to review these differences and work to further improve the drug development process.
This standard establishes recommendations for the planning of non-clinical safety studies for the purpose of subsequent clinical trials and registration of medicinal products.
This standard is applicable to all cases of drug development and provides general guidelines for drug development.
For medicinal products obtained using biotechnological methods, appropriate safety studies must be carried out in accordance with the ICH S6 guideline on non-clinical studies of biotechnological medicinal products. For these medicinal products, this standard applies only to the order of preclinical studies depending on the phase of clinical development.
To optimize and accelerate the development of drugs intended to treat life-threatening or serious diseases (for example, advanced cancer, persistent HIV infection, conditions caused by congenital enzyme deficiencies), for which there is currently no effective therapy, an individualized approach is also used as to toxicological evaluation and clinical development. In these cases, as well as for innovative therapeutics (eg, small interfering RNA) and vaccine adjuvants, certain studies may be shortened, modified, added, or deleted. If ICH guidelines are available for individual pharmacotherapeutic groups of medicinal products, the latter should be followed.
Drug development is a step-by-step process that involves evaluating data on its effectiveness and safety in both animals and humans. The primary goals of preclinical drug safety assessment include determining target organ toxicity, its dose-response relationship, its relationship to exposure (systemic exposure), and, if applicable, the potential reversibility of toxic effects. These data are used to determine the initial safe dose and dose range for clinical studies and to establish parameters for clinical monitoring of potential adverse effects. Preclinical safety studies, although limited in nature at the outset of clinical development, should be sufficient to indicate potential adverse effects that may occur in the planned clinical trial setting.
Clinical studies are conducted to study the effectiveness and safety of a drug, starting with relatively low systemic exposure in a small number of subjects. In subsequent clinical studies, drug exposure is increased by increasing the duration of use and/or the size of the study population. Clinical studies should be expanded with adequate evidence of safety based on the results of previously conducted clinical studies and based on additional nonclinical safety data that is obtained as clinical development progresses.
Clinical or preclinical data of serious adverse effects may influence the continuation of clinical studies. As part of the overall clinical development plan, these data should be reviewed to determine the feasibility of conducting and designing additional preclinical and/or clinical studies.
Clinical trials are conducted in phases, which have different names in different countries. This standard uses the terminology used in the ICH E8 guidance on general principles for the conduct of clinical trials of medicinal products. However, as there is a strong trend toward merging phases of clinical development, this document also, in some cases, also identifies the relationship of preclinical studies to the length and size of clinical studies, as well as the characteristics of the subjects involved (the target population).
The planning and design of nonclinical safety studies and human clinical trials should be based on scientific principles and consistent with ethical principles.
2.1 Selection of high doses for studying general toxicity
Potential clinically significant effects in toxicology studies can generally be fully studied at doses close to the maximum tolerated dose (MTD). However, it is not necessary to confirm MTD in every study. It is also permissible to use limited high doses, including doses that are multiples of the doses expected to be used in clinical practice (clinical exposure) or at which the maximum achievable exposure (saturation exposure) or acceptable maximum dose (MFD) is achieved. The use of these limited high doses (detailed below and in Figure 1) avoids administering doses to animals that do not provide additional information to predict clinical safety. This approach is consistent with similar recommendations for the design of reproductive toxicity and carcinogenicity studies that have already defined limited high doses and/or exposures.
A limited high dose of 1000 mg/kg/day for acute, subchronic and chronic toxicity studies in rodents and non-rodents is considered appropriate for all applications except those discussed below. In some cases, when a dose of 1000 mg/kg/day does not provide 10 times the clinical exposure, and the clinical dose of the drug exceeds 1 g/day, then doses in toxicology studies should be limited to 10 times the dose to achieve clinical exposure, the 2000 mg dose /kg/day or use MFD, choosing the smallest one. In those rare cases where the 2000 mg/kg/day dose is below clinical exposure, a higher dose up to the MFD may be used.
Doses that provide 50-fold excess systemic exposure (usually determined by the group mean AUC values (Note 1) of the parent substance or pharmacologically active prodrug molecule) compared with systemic clinical exposure are also considered acceptable maximum doses for acute and acute toxicity studies. repeated administration in any animal species.
To initiate phase III clinical trials in the United States, toxicology studies using limited high doses are conducted in at least one animal species at a dose that provides 50 times the exposure. If this approach is not feasible, it is recommended that the study be conducted in a single animal species for 1 month or more using a limited high dose of 1000 mg/kg, MFD or MTD, whichever is lower. However, in selected cases such a study may not be necessary if, in a shorter duration study, toxic effects were observed at doses greater than 50 times the exposure. If genotoxicity endpoints are included in a general toxicity study, then the appropriate maximum dose should be based on MFD, MTD, or a limited high dose of 1000 mg/kg/day.
Note 1—In this document, “exposure” generally refers to the mean AUC value in the group. In some cases (for example, if a compound or class of compounds is capable of causing acute cardiovascular changes or symptoms are associated with effects on the central nervous system), it is more appropriate to determine exposure limits by means of group mean C values.
Pharmacological and pharmacodynamic safety studies are defined in ICH Guideline S7A.
The main set of pharmacological safety studies includes assessment of effects on the cardiovascular, central nervous and respiratory systems. In general, these studies should be conducted prior to clinical development in accordance with the principles set out in ICH guidelines S7A and S7B for studying the pharmacological safety of medicinal products and for the preclinical assessment of the ability of medicinal products for human use to slow ventricular repolarization (prolong the QT interval). If necessary, additional and follow-up pharmacological safety studies may be conducted late in clinical development. To reduce the use of laboratory animals, other assessments should be included in general toxicity study protocols whenever possible. in vivo as additional.
The purpose of primary pharmacodynamic studies ( in vivo and/or in vitro) is to establish the mechanism of action and (or) pharmacological effects of the active substance in connection with its proposed therapeutic use. Such studies are typically conducted early in pharmaceutical development and are thus typically not conducted in accordance with the Principles of Good Laboratory Practice (GLP). The results of these studies can be used to guide dose selection for both preclinical and clinical studies.
Before commencing clinical trials, the metabolic profile and degree of plasma protein binding in animals and humans should generally be assessed. in vitro, as well as systemic exposure data (ICH S3A Guide to Toxicokinetic Studies) in animal species used in repeated dose toxicology studies. Pharmacokinetics (PK) data (i.e., absorption, distribution, metabolism, and excretion) of the species being studied should be obtained before clinical trials are initiated in large numbers of subjects or over an extended period of time (typically prior to the start of phase III clinical trials). animals and biochemical data obtained in vitro, significant for identifying potential drug interactions. These data are used to compare metabolites in humans and animals and determine the need for additional research.
Preclinical characterization of the metabolite(s) in humans is only necessary when exposure to the metabolite(s) exceeds 10% of the total drug exposure and the magnitude of exposure in humans is significantly greater than that observed in toxicological studies. Such studies must be conducted to obtain approval for phase III clinical trials. For drugs whose daily administered dose does not exceed 10 mg, such studies may be required at higher proportions of metabolites. Some metabolites are not subject to toxicological studies (eg, most methionine conjugates) and do not require study. The need for preclinical study of metabolites that may have possible toxicological effects (e.g., a metabolite unique to humans) must be considered on a case-by-case basis.
Traditionally, acute toxicity data have been obtained from single-dose toxicity studies in two mammalian species using clinically proposed and parenteral routes of administration. However, this information can also be obtained from properly conducted dose escalation studies or short-term dose range studies at which the MTD is determined for animals used in general toxicity studies.
Where acute toxicity information can be obtained from other studies, separate single-dose studies are not recommended. Studies providing information on acute toxicity may be limited to use only by the route of administration proposed for clinical use and may not be conducted in accordance with GLP requirements if the repeated dose toxicity studies conducted in accordance with GLP requirements used the route administration of a drug proposed for clinical use. Mortality should not be a mandatory end point in acute toxicity studies. In some special cases (eg, microdose studies, see Section 7), acute toxicity studies or single-dose studies may provide the primary rationale for conducting human clinical trials. In these cases, the choice of high dose may differ from that described in section 1.1, but should take into account the intended clinical dose and route of administration of the drug. These studies must be carried out in accordance with GLP requirements.
Information on the acute toxicity of drugs can be used to predict the consequences of overdose in humans and should be available before the start of phase III clinical trials. Earlier assessment of acute toxicity may be required for drugs proposed to treat patient populations at high risk of overdose (eg, depression, pain, dementia) in outpatient clinical trials.
The recommended duration of repeated dose toxicity studies depends on the duration, indication, and focus of the planned follow-up clinical study. In general, the duration of animal toxicity studies conducted in two animal species (one of which is nonrodent) should be equal to or greater than the planned duration of clinical studies, up to the recommended maximum duration of repeated dose toxicity studies (Table 1). The limited high doses/exposures considered suitable for repeated dose toxicity studies are described in 2.1.
In cases where a significant therapeutic effect is observed in clinical studies, the duration of the study may be increased on an individual basis compared to the duration of the repeated dose toxicity studies used as the basis for conducting clinical studies.
6.1 Research required for clinical development
In general, a multiple-dose toxicity study in two species (one of which is non-rodent) with a minimum duration of two weeks is sufficient to justify the feasibility of any clinical studies of up to two weeks' duration (Table 1). To justify clinical studies of longer duration, toxicity studies of at least the same duration are required. To justify clinical studies longer than 6 months, a 6-month rodent study and a 9-month nonrodent study are required (see notes to Table 1 for exceptions).
Table 1 - Recommended duration of repeated dose toxicology studies required to justify clinical trials
Maximum duration of a clinical study | ||
Rodents | Non-rodents |
|
Up to two weeks | 2 weeks | |
From two weeks to six months | Same as in clinical studies |
|
More than six months | 6 months | 9 months |
In the United States, the use of a single-dose extended toxicity study is permitted as an alternative to 2-week studies to support single-dose clinical trials (see note c in Table 3). Clinical studies of less than 14 days duration may be justified by toxicity studies of the same duration. In some cases, clinical studies of greater duration than 3 months may be initiated with results from 3 months of rodent and non-rodent studies, provided that the results of completed chronic toxicity studies in rodents and non-rodents in accordance with national regulatory requirements for clinical studies can be submitted before the clinical use of the medicinal product exceeds 3 months. For serious or life-threatening illnesses or on a case-by-case basis, such extension is possible subject to the availability of results from fully completed chronic toxicity studies in rodents and the results of intravital studies and necropsy data in non-rodent studies. Complete pathological data in nonrodents should be obtained within the next 3 months. There may be cases where the drug is intended for pediatric use, and available preclinical animal studies (toxicological or pharmacological) indicate a potential effect on the development of target organs. In these cases, long-term toxicity studies initiated in immature animals may be required (see section 12). In the European Union, toxicological studies of 6 months in non-rodents are considered sufficient. However, if studies of longer duration have been performed, additional studies beyond 6 months are not acceptable. The following are examples where non-rodent studies of 6 months duration are also suitable to support clinical trials in Japan and the USA: If immunogenicity or intolerance precludes long-term studies; For short-term exposure with repeated administration, even if the duration of the clinical study exceeds 6 months, for example with irregular use for migraine, erectile dysfunction or herpes simplex; Medicines used long-term to reduce the risk of cancer recurrence; Medicines used for indications for which a short life expectancy has been established. |
||
6.2 State registration
Considering the large number of patients at risk and the relatively less controlled conditions of drug use in medical practice, in contrast to clinical trials, to justify the possibility of medical use of a drug, preclinical studies of a longer duration are required than to justify clinical studies. The duration of repeated dose toxicity studies required to justify the approval for medical use of drugs with different treatment durations is given in Table 2. In some cases, for a small number of pathological conditions, when the recommended duration of drug use is from 2 weeks to 3 months, but there is a large Clinical experience suggesting broader and longer-term clinical use (eg, anxiety, seasonal allergic rhinitis, pain), toxicology studies with a duration more appropriate for cases where the recommended duration of drug use exceeds 3 months may be required.
Table 2 - Recommended duration of toxicological studies with repeated administration required for state registration of a medicinal product*
Duration of use according to indication | Non-rodents |
|
Up to two weeks | ||
Over two weeks to one month | ||
Over one month to three months | 6 months | 6 months |
Over three months | 6 months | 9 months |
* Explanations are provided in the notes to Table 1. |
||
Determining the dose to be administered to humans for the first time is an important element in ensuring the safety of subjects participating in early clinical trials. When determining the recommended starting dose for humans, all relevant nonclinical data should be evaluated, including pharmacological dose-response effects, pharmacological/toxicological profile, and pharmacokinetic data.
In general, the most important information is provided by the high nontoxic dose (NOAEL) established in nonclinical safety studies in the most appropriate animal species. The estimated clinical starting dose may also depend on various factors, including pharmacodynamic parameters, individual properties of the active substance, and the design of clinical studies. Selected approaches are presented in national guidelines.
Exploratory clinical studies (Section 8) in humans may be initiated with less or a different volume of nonclinical studies than required for clinical development studies (6.1), and therefore the determination of the clinical starting (and maximum) dose may be different. Recommended criteria for the selection of starting doses in various exploratory studies are given in Table 3.
In some cases, the availability of early human data can provide a better understanding of the physiological/pharmacological characteristics of a drug in humans, the properties of the drug under development, and the relevance of therapeutic targets to a given disease. Rational early exploratory research can solve such problems. For the purposes of this standard, exploratory clinical trials are defined as studies conducted early in phase I, involving limited exposure and not assessing therapeutic efficacy and clinical tolerability. They are carried out to study various parameters such as PD, PK of the drug and other biomarkers, which may include receptor binding and displacement determined by PET, or other diagnostic parameters. The subjects of these studies can be either patients from the target population or healthy volunteers.
In these cases, the extent and type of nonclinical data required will depend on the magnitude of human exposure, taking into account the maximum clinical dose and duration of use. Five different examples of exploratory clinical trials are grouped and described in more detail below and in Table 3, including preclinical research programs that may be recommended in these cases. It is also possible to use alternative approaches not described in this standard, including approaches to justify clinical trials of biotechnological medicinal products. Alternative approaches to exploratory clinical trials are recommended to be discussed and agreed with the appropriate regulatory authorities. Any of these approaches could lead to an overall reduction in the use of laboratory animals in drug development.
Recommended starting doses and maximum doses for use in toxicology studies are given in Table 3. In all cases, establishment of PD and pharmacological parameters using models in vivo and/or in vitro is extremely important, as indicated in Table 3 and Section 2, and these data should be used to justify the selected human dose.
8.1 Clinical studies using microdose
The two different microdose approaches presented in this section are described in more detail in Table 3.
In the first approach, the total dose of the drug should be no more than 100 mcg, which is administered to each study subject simultaneously (one dose) or in several doses. The study is carried out to study the binding of target receptors or the distribution of a substance in tissues using PET. Also, the purpose of such a study may be to study PK with or without the use of a radioactive label.
In the second approach, study subjects are given 5 or fewer doses of no more than 100 mg (for a total of 500 mcg per subject). Such studies are carried out with similar objectives as using the above approach, but in the presence of less active PET ligands.
In some cases, it may be appropriate to conduct a clinical trial using microdoses and intravenous administration of a drug intended for oral administration, with full preclinical toxicological data available for the oral route. However, the intravenous microdose may be considered, based on the availability of toxicological data for the oral route, as described in Tables 1 and 3, as approach 3, in which acceptable exposure levels were achieved. In this case, it is not recommended to study intravenous local tolerance of the active substance, since the administered dose is extremely low (no more than 100 mcg). If a new diluent is used in an intravenously administered drug, the local tolerability of the diluent should be studied.
8.2 Single-dose clinical studies in the subtherapeutic range or expected therapeutic range
In this approach (Approach 3), a single-dose clinical trial is conducted, typically starting at subtherapeutic doses and subsequently increasing to the pharmacologically effective or expected therapeutic range (see Table 3). The determination of the acceptable maximum dose should be based on nonclinical data, but it may be further limited based on clinical data obtained during the ongoing study. The use of this approach may allow, for example, to determine PK parameters with the administration of a drug without a radioactive label at a predicted pharmacodynamically effective dose or close to it. Another example of the application of this approach is the assessment of on-target action or pharmacological action after a single administration. Studies using this approach are not designed to support the maximum tolerated clinical dose (see exceptions, note "a" in Table 1).
8.3 Clinical studies using multiple doses
To justify clinical studies using multiple doses, two different approaches are used for preclinical studies (Approaches 4 and 5 in Table 3). Studies based on them can justify the duration of administration of drugs in doses of the therapeutic range for 14 days to assess PK and PD parameters in humans, but they are not used to justify the determination of the tolerated maximum clinical dose.
Approach 4 involves a two-week, multiple-dose toxicology study in rodents and non-rodents. The choice of dosage administered to animals is based on the multiple exposure dose at the expected AUC at the maximum clinical dose.
Approach 5 involves a two-week toxicology study in rodents and a confirmatory toxicology study in non-rodents to confirm that the NOAEL is not toxic to rodents when administered to non-rodents. If a toxic effect is observed in rodents when administered to non-rodents, clinical use of the drug should be delayed until data from subsequent non-clinical studies in animals of that species (usually a standard toxicology study, section 5) are available.
Table 3 - Recommended preclinical studies to justify the possibility of conducting exploratory clinical studies
Clinical researches | Preclinical studies |
|||
Doses administered | Initial and maximum doses | Pharmacology | General toxicity studies | Study of genotoxic |
Total dose 100 mcg (no dosing interval), and total dose 1/100th NOAEL and 1/100th pharmacological | Initial and maximum doses may be the same, but should not exceed a total dose of 100 mcg | Target/receptor profile in vitro must be appreciated | Extended single-dose toxicology study (see notes c and d) in a single animal species, typically rodents, using a proposed route of administration for clinical use to obtain toxicokinetic effects. | For effective radioactive labels (for example, labels for PET), appropriate |
The total cumulative dose is 500 mcg, no more than 5 administrations of the drug with a washout period between administrations (6 or more actual or predicted | The initial and maximum doses can be the same, but should not exceed 100 mcg | Target/receptor profile in vitro must be appreciated To justify the choice of dose for use in humans, detailed data on the main (primary) pharmacological parameters (mechanism of action and/or effects) must be obtained using a pharmacologically relevant model | A toxicological study lasting 7 days with repeated administration to animals of the same species, usually rodents, using the route of administration proposed for clinical use to obtain toxicokinetic Hematological, clinical laboratory, necropsy and histopathological data must be obtained A maximum dose of 1000 times the clinical dose, converted to mg/kg for IV administration and mg/m2 for oral administration, can be used. | A genotoxicity study is not required, but any SAR studies or assessments performed must be included in the clinical trial authorization documents. For effective radioactive tracers (e.g. PET tracers), appropriate PK estimates of tracer parameters and dosimetric data should be provided |
Single dose subtherapeutic studies | The choice of initial starting dose should be based on the types of toxicological data obtained from the most sensitive laboratory animal species and data on the pharmacologically effective dose. National recommendations for the selection of the initial starting dose for humans should also be taken into account. The maximum dose may be set at up to 1/2 the NOAEL of exposure in the most sensitive species of laboratory animals in cases where any significant toxic effect observed in animals is possible and reversible in humans | Target/receptor profile in vitro must be appreciated To justify the choice of dose for use in humans, detailed data on the main (primary) pharmacological parameters (mechanism of action and/or effects) must be obtained using a pharmacologically relevant model. Core set of pharmacological safety studies (see section 2) | Extended single-dose toxicology study (see notes c) along the intended clinical route of drug administration, obtaining toxicokinetic, hematological, laboratory clinical data, necropsy data and histopathological examination. In this case, MTD, MFD or limited high dose is used as the high dose (see 1.1) |
|
Administration of the drug for 14 days in therapeutic | If toxic effects occur in both types of laboratory animals, national requirements for the selection of the initial clinical dose should be followed. If toxic effects were not observed in any laboratory animal species (i.e., NOAELs represent the highest doses tested in preclinical studies and the doses used were not restricted in any way, such as not representing MFD) or were observed in only one laboratory animal species, then the initial clinical the dose should be one of the doses that achieves the predicted clinical AUC (based on either cross-species PK modeling or mg/m2 conversion) that is 1/50th the AUC of NOAEL in animals and at which the lower exposure was obtained In the absence of toxic effects in both species, it is recommended to use a maximum clinical dose not exceeding 1/10th of the lower exposure (AUC) in either species obtained in either species at the highest dose. If toxic effects are observed in only one species of animal, the maximum clinical dose should not exceed the NOAEL for the species in which toxic effects were observed or 1/2 the AUC of the highest administered dose at which toxic effects were absent (whichever is lower). ). If toxic effects are present in both animal species, the choice of the maximum clinical dose should be based on a standard risk assessment approach, and in this special case the clinical MTD may be assessed. | Target/receptor profile in vitro must be appreciated To justify the choice of dose for use in humans, detailed data on the main (primary) pharmacological parameters (mechanism of action and/or effects) must be obtained using a pharmacologically relevant model. A core set of pharmacological safety studies (see section 2) using doses similar to those in general toxicology | Toxicological study lasting 14 days with repeated administration in rodents and non-rodents with a standard set of evaluated parameters; the choice of dose used is based on the multiple exposure of the expected clinical AUC at the maximum dose | The Ames test (or an alternative test if the Ames test is not acceptable, for example for antibacterial |
Administration of the drug within 14 days, without exceeding the duration | Predicted exposure when administering starting doses should not exceed 1/50th of the NOAEL in the most sensitive animal species, calculated as mg/m. National recommendations for the selection of initial clinical dose should be taken into account Maximum exposure in humans should not exceed the AUC at NOAEL in nonrodents or 1/2 the AUC at NOAEL in rodents, whichever is less. | Target/receptor profile in vitro must be appreciated To justify the choice of dose for use in humans, detailed data on the main (primary) pharmacological parameters (mechanism of action and/or effects) must be obtained using a pharmacologically relevant model. Core set of pharmacological safety studies (see section 2) using doses similar to those in general toxicology studies | A standard 14-day repeated dose toxicology study in rodents (with justification for selecting rodents as an acceptable laboratory animal species for this study). The high dose is MTD, MFD or limited high dose (see 1.1) Confirmatory study in non-rodents n=3) at an expected NOAEL rodent exposure of at least 3 days and the shortest duration of the intended clinical trial An alternative non-rodent dose escalation study of at least 3 days duration and the shortest duration of the intended clinical study at dose administration may be conducted to achieve the NOAEL exposure in rodents. | The Ames test (or an alternative test if the Ames test is not acceptable, for example for antibacterial |
General toxicity preclinical studies must be conducted in accordance with GLP rules. Genotoxicity study design and dose selection are described in ICH guidance S2B. The design of a single-dose extended study should typically include evaluation of hematologic, laboratory, clinical, necropsy, and histopathologic data (only control and high doses are administered if drug toxicity is not observed with the high dose) after a single dose, followed by observation for two weeks to assess delayed toxic effects and/or their resolution. The standard rodent study design includes toxicological evaluation of 10 animals/sex/group one day after drug administration, with 5 animals/sex receiving selected dose(s) assessed on day 14 post-dose. The standard non-rodent study design includes 3 animals/sex/group assessments for all groups on day 2 post-dose and 2 animals/sex assigned to the selected dose(s) assessed on day 14 post-dose. A single dose level to assess reversibility/delay of toxic effects at 14 days post-dose can be used to justify a microdose approach. The dose level used to administer to the animal should not be set at a high dose level, but should be at least 100 times the clinical dose. In the absence of adverse effects in clinical trials, dose escalation above this AUC may be acceptable if toxicological data suggest that potential adverse effects in humans are detectable, reversible, and of low severity. |
||||
Local tolerability with the proposed method of administration in clinical studies is preferably studied as part of a study of general toxicity; Individual studies are usually not recommended.
To justify limited clinical studies of an alternative therapeutic route of administration (for example, a single intravenous administration to determine the absolute bioavailability of a drug taken orally), a study of the tolerability of a single dose in a single animal species is acceptable. In cases where the expected systemic exposure (AUC and Cmax) for a non-therapeutic route of administration has been studied through established toxicology studies, local tolerability study endpoints may be limited to clinical effects and macro- and microscopic examination of the site of administration. The composition of the medicinal product intended for local tolerability studies may not be identical, but should be similar to the composition and dosage form of the medicinal product used in the clinical trials.
For IV microdose studies, which are conducted when oral toxicology data are available (see section 7), an assessment of local tolerance of the pharmaceutical substance is not required. If a new solvent is used in the composition of the drug for intravenous administration, it is necessary to study its local tolerability.
For parenteral medicinal products, studies of local tolerability at unintended injection sites, if required, should be conducted before prescribing the medicinal product to a large number of patients (for example, before phase III clinical trials). The approach to planning such studies varies from country to country. Such studies are not required in the United States (an example exception would be intrathecal administration when epidural administration is planned). In Japan and EU countries, a single paravenous injection is recommended for the IV route. The need to explore other parenteral routes of administration is assessed individually.
Testing for gene mutations is considered sufficient to support all single-dose clinical trials. Additional studies to identify chromosomal damage in mammals are needed to support multiple-dose clinical trials. A full battery of genotoxicity tests must be performed prior to the start of phase II clinical trials.
If the results of the study indicate the presence of genotoxic effects, it is necessary to evaluate them and, possibly, conduct additional studies to establish the acceptability of further use of the drug in humans.
Genotoxicity studies recommended to support exploratory clinical studies using different approaches are discussed in Section 8 of this standard.
Cases requiring carcinogenicity studies are discussed in ICH S1A guidance on assessing the need for carcinogenicity studies of medicinal products. In these cases, carcinogenicity studies must be carried out before the start of the state registration procedure. In cases where there are compelling reasons indicating a carcinogenic risk, the results of the studies must be presented before clinical trials are carried out. The long duration of a clinical study is not considered a mandatory reason for carcinogenicity studies.
Necessary studies of the carcinogenicity of drugs developed for the treatment of serious diseases in adults and children may be carried out, in agreement with the regulatory authority, after their state registration.
Reproductive toxicity studies should be conducted taking into account the patient population that will use the study drug.
12.1 Men
Males may be included in phase I and phase II clinical studies prior to male reproductive evaluation due to the fact that male reproductive evaluation is performed in repeated dose toxicity studies.
Note 2—Assessment of male and female fertility by standard histological examination of the testes and ovaries in toxicity studies (with repeated administration, usually in rodents) of at least 2 weeks duration is considered comparable in ability to detect toxic effects to fertility studies to detect toxic effects on the reproductive organs of males and females
Fertility studies in males should be completed before large-scale or long-term clinical studies (eg, phase III studies) are initiated.
12.2 Women without childbearing potential
If appropriate repeated dose toxicity studies have been conducted (which include assessment of female reproductive organs), it is permissible to include women without reproductive potential (i.e., permanently sterilized, postmenopausal) in clinical studies without reproductive toxicity studies. Postmenopause is defined as the absence of menstruation for 12 months without other medical reasons.
12.3 Women of childbearing potential
For women of childbearing potential (WOCBP), there is a high risk of unintentional exposure of the embryo or fetus to the drug before information about the potential benefit and risk is known. All countries using ICH guidelines have similar recommendations regarding the timing of reproductive toxicity studies for the inclusion of WSDP in clinical trials.
When including WSDP in studies, the risk of unintended exposure to the embryo or fetus must be identified and minimized. The first approach to achieving this goal is to conduct reproductive toxicity studies to assess the risk of the drug and take appropriate precautions in clinical trials in women with diabetes. The second approach is to limit risks by taking precautions to prevent pregnancy during clinical trials. These measures include pregnancy tests (eg, free (3-subunit) hCG), use of highly reliable methods of contraception (Note 3), and enrollment in the study only after confirmation of menses. Clinical trial pregnancy tests and patient education should be sufficient to ensure that measures aimed at preventing pregnancy are implemented during the period of drug exposure (which may exceed the duration of the study).To ensure these approaches, informed consent should be based on all available information on reproductive toxicity, such as: a general assessment of the potential toxicity of the drug drugs with similar structure or pharmacological effects.If significant information on the effect on reproduction is not available, the patient must be informed of the potential unidentified risk to the embryo or fetus.
In all countries that apply ICH guidelines, under certain conditions, it is permissible to include VSD in early phase clinical trials without preclinical developmental toxicity studies (e.g. without studies of possible effects on embryonic and fetal development). One such requirement is that the risk of pregnancy be adequately monitored during short-term (eg, 2-week) clinical studies. Another condition may be the predominance of the disease among women, when it is impossible to achieve the goal of the study without including WSDP, while sufficient measures have been taken to prevent pregnancy (see above).
Note 3—Highly reliable methods of contraception are considered to be both single and combination drugs that provide a low pregnancy rate (i.e. less than 1% per year) when used consistently and correctly. For patients using hormonal contraceptives, information about the effect of the study drug on contraception must be provided.
Additional justification for conducting studies in WSD without preclinical developmental toxicity studies is knowledge of the mechanism of action of the drug, its properties, duration of fetal exposure, or the difficulty of conducting developmental toxicity studies in a suitable animal model. For example, for monoclonal antibodies, which, according to current scientific data, have weak embryonic and fetal effects during organogenesis, developmental toxicity studies can be conducted during phase III clinical trials. A report of the completed study must be submitted as part of the registration dossier.
In general, if there is preliminary data on reproductive toxicity in two animal species (Note 4) and if measures to prevent pregnancy are in place (see above), inclusion of WSDP (up to 150 subjects) receiving the investigational medicinal product for a relatively short period (up to 3 months) until specific reproductive toxicity studies are conducted. The rationale for this is the very low pregnancy rates in controlled studies of this size and duration (Note 5) and the ability of well-designed pilot studies to identify the most important developmental toxicities that may highlight the risks of including WSDP in clinical trials. The number of women who are included in the study and the duration of the study may be influenced by population characteristics that reduce the likelihood of pregnancy (eg, age, disease).
Note 4—If dosages are adequate, a preliminary embryonic and fetal development study, which includes assessment of fetal survival, body weight, external and internal organ studies, using at least six females per group and including females treated with the medicinal product, is appropriate to achieve this goal. period of organogenesis. Such preliminary preclinical studies must be conducted to high scientific standards with easy access to data or in accordance with GLP requirements.
Note 5—The pregnancy rate in women trying to conceive for the first time is approximately 17% per menstrual cycle. The pregnancy rate in phase III studies conducted in women of childbearing potential is<0,1% на менструальный цикл. В ходе этих исследований пациентов следует предупредить о нежелательности наступления беременности и необходимости соблюдения мер по предупреждению беременности. По имеющимся данным, частота наступления беременности во II фазе ниже, чем в III фазе, но в силу ограниченного количества включенных женщин величину снижения установить невозможно. Основываясь на данных III фазы, частота наступления беременности во II фазе исследований, включающих 150 женщин с сохраненным детородным потенциалом и продолжительностью до 3 месяцев, значительно меньше 0,5 беременностей на лекарственный препарат, находящийся в разработке.
In the United States, studies of embryonic and fetal development may be delayed until phase III studies with the inclusion of VSD while taking measures to prevent pregnancy (see above). In the EU and Japan (except for the situations described above in this section), specific developmental toxicity studies must be completed before inclusion of WSDP in the study.
In all countries implementing ICH guidelines, it is permissible to include WSDP in phase I and II multiple dose clinical trials prior to female fertility studies, given that animal reproductive system assessment is carried out as part of multiple dose toxicity studies (note 2). Dedicated preclinical female fertility studies are required to include WSDP in large-scale, long-term clinical trials (e.g., phase III studies).
In all countries that apply ICH guidelines, the results of pre- and postnatal otgenetic development studies must be submitted for state registration of a medicinal product.
Submission of data from a completed reproductive toxicity study and a standardized battery of genotoxicity tests are required prior to inclusion in any studies of WHDCs who are not using highly effective methods of contraception (Note 3) or of unknown gestational status.
12.4 Pregnant women
A complete reproductive toxicity study and a standardized battery of genotoxicity tests should be performed before pregnant women are included in clinical trials. In addition, it is necessary to evaluate the available data on the safety of the drug in humans.
When justifying the inclusion of pediatric patients in clinical trials, the most relevant information is safety data from previous studies in adult patients and should be available before studies in children are initiated. The sufficiency and extent of clinical trial data in adults to support this decision is determined on a case-by-case basis. Prior to use in children, sufficient data on experience in adults may not be available (for example, with exclusively pediatric indications for use).
Prior to initiation of studies in children, the results of repeated dose toxicity studies of appropriate duration in adult animals (see Table 1), a core set of pharmacological safety studies, and a standard set of genotoxicity tests should be completed. Reproductive toxicity data appropriate for the age and sex of the children studied, providing information on direct toxic risk or developmental effects (eg, fertility studies, pre- and postnatal development) may also be needed. Embryonic and fetal development studies are not critical to justify clinical trials in male or prepubertal female patients.
The need for any studies in immature animals should only be considered if previous animal and human safety data, including the effects of other medicinal products in the same pharmacological class, are considered insufficient to justify a clinical trial in children. If such preclinical research is necessary, the use of a single animal species, preferably rodents, is sufficient. Subject to sufficient scientific justification, research on non-rodents is permitted.
For short-term PK studies in children (eg, 1-3 doses), toxicity studies in immature animals are generally not considered informative.
Depending on the indication for use, the age of children included in the clinical trial, and safety data for adult animals and patients, consideration should be given to the need to obtain results from studies in immature animals before initiating short-term clinical studies of efficacy using a wide range of doses and the safety of the drug. drug. One of the most important issues is the age of study participants in relation to study duration (that is, the proportion of the developmental period during which study participants take the drug). This factor is decisive in assessing the need to conduct preclinical studies on immature animals, and, if they are necessary, the timing of their conduct in relation to clinical studies should be established.
Before long-term clinical studies in pediatric patients that require toxicity studies in immature animals are initiated, these preclinical studies must be completed.
There may be situations where pediatric patients are the primary therapeutic population and available experimental data indicate a potential effect of the study drug on target organ development (toxicological or pharmacological). In some of these cases, long-term studies in immature animals may be required. A long-term toxicological study in animals of the appropriate species and age is acceptable (eg, a 12-month study in dogs or a 6-month study in rats). A 12-month study can cover the entire developmental period in dogs. For other laboratory animal species, this design can be adapted to replace the appropriate standard chronic study and a separate study in immature animals under certain conditions.
Before long-term clinical studies in children are initiated, the need for carcinogenicity studies must be determined. However, if there are no significant reasons (for example, evidence of hepatotoxicity in various tests or the presence of a pro-carcinogenic risk due to the mechanism of action or effects identified in the study of general toxicity) there is no need to study carcinogenicity for clinical trials in children.
As stated in ICH S8 guidance on drug immunotoxicity studies, all new drug products should be assessed for immunotoxic potential using standard toxicology studies and additional immunotoxicity studies conducted based on a review of the totality of evidence, including immune-mediated signals identified in standard toxicology studies. If there is a need to conduct additional immunotoxicity studies, they should be completed before using the investigational medicinal product in larger patient populations (eg, phase III clinical studies).
The need or timing of a photosafety study depending on a person’s exposure is determined by:
- photochemical properties (for example, photoabsorption and photostability) of the molecule;
- information on the phototoxic potential of chemically similar compounds;
- distribution in tissues;
- clinical or preclinical data indicating the presence of phototoxicity.
An initial assessment of phototoxic potential should be made based on the photochemical properties of the drug and its pharmacological/chemical class. If an assessment of all available data and the proposed clinical trial design indicates a significant risk of phototoxicity to humans, patient protection measures should be in place during outpatient clinical trials. In addition, subsequent nonclinical evaluation of the distribution of the active substance in the skin and eyes is necessary to provide information about the risk to humans and the need for further study. Then, if applicable, experimental evaluation (preclinical, in vitro or in vivo, or clinical) phototoxic potential should be carried out before using the drug in a large number of patients (phase III clinical studies).
Alternatively, instead of the stepwise approach described above, direct assessment of phototoxic potential can be performed in preclinical or clinical studies. If the results of these studies are negative, then early assessment of drug distribution in the eyes/skin and preventive measures during the clinical trial are not required.
If phototoxicity evaluation results indicate possible photocarcinogenic potential, the risk is usually adequately controlled in patients with protective measures including a warning in the informed consent and instructions for use (see Note 6).
Note 6—Studying photocarcinogenicity in non-rodents using currently available models (eg, hairless rodents) for drug development is considered impractical and is generally not required. If phototoxicity studies indicate a possible photocarcinogenic risk and an appropriate test method becomes available, the study usually needs to be completed before the registration process is initiated and the results must be taken into account in assessing the risk to humans.
For drugs that affect the central nervous system, regardless of indications for use, it is necessary to determine the need to assess the risk of developing drug dependence. Preclinical studies are necessary to justify the design of clinical trials, determine a special category used in the country (for example, lists of narcotic and psychotropic substances, etc.), and draw up instructions for use. When forming a set of necessary studies, one should be guided by national guidelines on preclinical assessment and the risk of developing drug dependence.
Preclinical data collected early in drug development can be informative in identifying early indicators of addiction potential. Data on such early indicators should be obtained prior to the first use of the drug in humans; these include a PK/PD profile to determine duration of action, similarity of chemical structure to drugs of abuse, receptor binding profile, and behavioral/clinical symptoms from preclinical studies in vivo. If these early studies do not reveal potential for drug dependence, extensive preclinical studies in models of drug dependence may not be necessary. In general, if an active substance exhibits features similar to known patterns of drug dependence or has a novel mechanism of action on the central nervous system, further preclinical studies are recommended before initiation of large clinical trials (eg, phase III clinical trials).
If the profile of metabolites and the target of action of the drug in rodents corresponds to those in humans, a nonclinical assessment of the risk of drug dependence is carried out in rodents. Non-human primates should only be used in rare cases where there is compelling evidence that such studies will predict human susceptibility to drug dependence and rodent models are inadequate. To assess the risk of developing drug dependence, three types of studies are most often conducted: drug preference, drug self-administration, and post-drug withdrawal assessment. Drug preference and self-administration studies are usually conducted as separate experiments. Withdrawal studies may sometimes be included in a repeated dose toxicity study (toxicity reversibility panel). The maximum dose that achieves plasma concentrations in laboratory animals several times higher than the therapeutic clinical dose in humans is considered appropriate for such nonclinical assessments of the risk of drug dependence.
If prior preclinical or clinical data on a drug or related drugs indicate the potential for specific safety concerns, additional preclinical studies (eg, to identify potential biomarkers, to elucidate mechanism of action) may be required.
ICH Guidelines Q3A and Q3B provide approaches to qualifying impurities and degradation products of the active substance. If special studies are required to qualify impurities and degradation products, they are generally not required until the start of Phase III clinical trials, unless development changes result in an essentially new impurity profile (e.g., new synthesis routes). , new degradation products formed as a result of interactions between the components of the drug). In such cases, appropriate impurity and degradation product qualification studies may be required to support Phase II or later developmental clinical studies.
This section applies to combination medicinal products that are intended for simultaneous use and are included in one package or for administration in one dosage form (“fixed combination”). The principles set out below can also be applied to non-combined medicinal products, which, according to the instructions for use, can be used simultaneously with a certain medicinal product, including not in the form of a “fixed combination”, as well as for medicinal products that do not have sufficient clinical data on the combination application.
This standard applies to the following combinations:
1) two or more substances in late stages of development (compounds with significant experience in clinical use (i.e. phase III clinical trials or post-registration studies);
2) one or more substances in late stages of development and one or more substances in early stages of development (there is limited clinical experience, such as a phase II clinical trial and earlier phase studies), or
3) more than one substance in the early stages of development.
For most combinations containing two substances that are in late stages of development but for which there is no significant clinical experience with joint use, combination toxicology studies are not required to justify clinical trials or regulatory approval unless there is reason to suspect a possible joint toxicological effect (e.g. , the presence of target organs identical for the toxic effect). These reasons may vary depending on the level of safety and the ability to monitor adverse effects in humans. If a nonclinical study is required to evaluate the possible additive toxicological effects of a combination, it should be completed before clinical trials of the combination are initiated.
For combinations containing two substances that are in late stages of development but for which there is no acceptable clinical experience with coadministration, preclinical studies of the combination are generally not available to support relatively short-term clinical studies (e.g., phase II studies of up to 3 months) are required if the opinion that there are no possible toxicological effects of the combination is based on sufficient available data. However, for long-term and large-scale clinical studies, as well as for the state registration procedure, preclinical studies of such combinations are mandatory.
For combinations of substances in early stages of development with clinical experience with substances in later stages of development for which there are no significant toxicological concerns, toxicological studies of the combination are not required to justify the feasibility of “proof of concept” studies of up to 1 month duration. . Clinical studies of the combination should not exceed clinical experience with the individual components in duration. For later stage and longer clinical trials, preclinical combination studies are required.
For combinations containing substances in the early stages of development, preclinical studies of their combination are necessary to justify the possibility of conducting clinical trials.
If a full preclinical research program has been conducted for each of the components of the combination, and preclinical toxicological studies of the combination are necessary to justify the feasibility of conducting a clinical trial, the duration of the combination study should be equivalent to the duration of the clinical trial (but not more than 90 days). This preclinical study will also be suitable for the state registration procedure. A preclinical study of a combination of shorter duration may also be eligible for regulatory approval depending on the duration of the intended clinical use.
The design of nonclinical studies recommended for studying a combination depends on the pharmacological, toxicological and pharmacokinetic profiles of the individual components, indications for use, the proposed target patient population and available clinical data.
Preclinical studies of the combination are usually conducted in one suitable animal species. If unexpected toxic effects are detected, additional studies may be required.
In cases where a complete nonclinical testing program has not been completed for the individual components, a complete nonclinical toxicology program may be conducted for the combination only, provided that the individual components are intended for use in the combination only.
If the individual components have been studied in accordance with current standards, then genotoxicity, pharmacological safety and carcinogenicity studies of the combination are usually not required for clinical trials or state registration procedures. In cases where the patient population includes WSDP and studies of the individual component(s) indicate embryonic and fetal risk, combination studies are not recommended because the potential for harm to human embryonic and fetal development has already been established. If preclinical studies of embryonic and fetal development indicate that neither component poses a risk to human developmental development, combination studies are not required unless there is concern, based on the properties of the individual components, that the combination may pose a safety risk to humans. In cases where the effect of individual components of the composition on embryonic and fetal development has been studied, but combination studies are required, the results of the latter must be submitted at the beginning of the state registration procedure.
Abbreviations
Area Under the Curve | Area under the pharmacokinetic curve |
|
Maximum Plasma Concentration | Maximum plasma concentration |
|
European Union |
||
Good Laboratory Practices | Good laboratory practice |
|
Human Chorionic Gonadotropin | Human chorionic gonadotropin |
|
Human Immunodeficiency Virus | Immunodeficiency virus |
|
International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use | International Conference on Harmonization of Technical Requirements for Registration of Medicines for Medical Use |
|
Intravenous |
||
Maximum Feasible Dose | Maximum permissible dose |
|
Maximum Tolerated Dose | Maximum Tolerated Dose |
|
VNTD (NOAEL) | No Observed Adverse Effect Level | High non-toxic dose |
Positron Emission Tomography | Positron emission tomography |
|
Pharmacokinetics | Pharmacokinetics |
|
Pharmacodynamics | Pharmacodynamics |
|
Structure-Activity Relationship | Relationships caused by the activity of a molecular structure |
|
Small Interfering RNA | Small interfering RNA |
|
WSDP (WOCBP) | Women of Childbearing Potential | Women of childbearing potential |
ICH S6 Guideline: Preclinical Safety Evaluation for Biotechnological-Derived Pharmaceuticals; July 1997.
ICH E8 Guideline: General Considerations for Clinical Trials; July 1997.
ICH S5(R2) Guideline: Detection of Toxicity to Reproduction for Medicinal Products and Toxicity to Male Fertility; June 1993.
ICH S1 C(R2) Guideline: Dose Selection for Carcinogenicity Studies of Pharmaceuticals; March 2008.
ICH S7A Guideline: Safety Pharmacology Studies for Human Pharmaceuticals; November 2000.
ICH S7B Guideline: The Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) By Human Pharmaceuticals; May 2005.
ICH S3A Guideline: Note for Guidance on Toxicokinetics: The Assessment of Systemic Exposure in Toxicity Studies; October 1994.
National Center for the Replacement, Refinement and Reduction of Animals in Research. Challenging Requirements for Acute Toxicity Studies: Workshop Report; May 2007.
Robinson S., Delongeas JL., Donald E., Dreher D., Festag M., Kervyn S. et al. A European pharmaceutical company initiative challenging the regulatory requirement for acute toxicity studies in pharmaceutical drug development. Regul Toxicol Pharmacol 2008;50:345-352.
ICH S2B Guideline: Genotoxicity: A Standard Battery for Genotoxicity Testing for Pharmaceuticals; July 1997.
ICH S1A Guideline: Guideline on the Need for Carcinogenicity Studies of Pharmaceuticals; November 1995.
ICH Q3A(R2) Guideline: Impurities in New Drug Substances; October 2006.
ICH Q3B(R2) Guideline: Impurities in New Drug Products; June 2006.
ICH S8 Guideline: Immunotoxicity Studies for Human Pharmaceuticals; September 2005.
Sakai T., Takahashi M., Mitsumori K., Yasuhara K., Kawashima K., Mayahara H. et al. Collaborative work to evaluate toxicity on male reproductive organs by 2-week repeated-dose toxicity studies in rats. Overview of the studies. J Toxicol Sci 2000;25:1-21.
Sanbuissho A., Yoshida M., Hisada S., Sagami F., Kudo S., Kumazawa T. et al. Collaborative work on evaluation of ovarian toxicity by repeated-dose and fertility studies in female rats. J Toxicol Sci 2009;34:1-22.
Application YES
(informative)
Table DA.1
Designation of the referenced international document | Degree of compliance | Designation and name of the corresponding national standard |
ICH S3A Guideline | GOST R 56702-2015 "Medicines for medical use. Preclinical toxicological and pharmacokinetic safety studies" |
|
ICH S6 Guideline "Medicinal products for human use. Non-clinical pharmacological safety studies" |
||
OECD Principles of good laboratory practice | GOST R 53434-2009 "Principles of Good Laboratory Practice" |
|
Note - This table uses the following conventions for the degree of compliance with standards: IDT - identical standards; MOD - modified standards. |
||
UDC 615.038:615.012/.014:615.2:006.354 | |
Key words: drugs, preclinical safety studies, clinical studies, state registration, safety |
|
Electronic document text
prepared by Kodeks JSC and verified against:
official publication
M.: Standartinform, 2016
Clinical trial (CT) - This is the study of the clinical, pharmacological, pharmacodynamic properties of the drug being studied in humans, including the processes of absorption, distribution, change and excretion, with the aim of obtaining, using scientific methods, assessments and evidence of the effectiveness and safety of drugs, data on expected side effects and interaction effects with other drugs.
The purpose of clinical trials of drugs is to obtain, by scientific methods, assessments and evidence of the effectiveness and safety of medicines, data on expected side effects from the use of medicines and the effects of interaction with other medicines.
In the process of clinical trials of new pharmacological agents, 4 interconnected phases:
1. Determine the safety of drugs and establish the range of tolerated doses. The study is carried out on healthy male volunteers, in exceptional cases - on patients.
2. Determine the effectiveness and tolerability of drugs. The minimum effective dose is selected, the breadth of therapeutic action and the maintenance dose are determined. The study is carried out on patients of the nosology for which the drug under study is intended (50-300 persons).
3. The effectiveness and safety of the drug, its interaction with other drugs in comparison with standard treatment methods are clarified. The study is carried out on a large number of patients (thousands of patients), with the involvement of special groups of patients.
4. Post-registration marketing studies study the toxic effects of the drug during long-term use and identify rare side effects. Different groups of patients can be included in the study - by age, according to new indications.
Types of clinical studies:
Open, when all trial participants know which drug the patient is receiving;
Simple “blind” - the patient does not know, but the researcher knows what treatment was prescribed;
In double-blind, neither the research staff nor the patient knows whether he is receiving the drug or a placebo;
Triple-blind - neither the research staff, nor the examiner, nor the patient knows which drug is being treated.
One of the types of clinical trials is bioequivalence studies. This is the main type of control of generic medicines that do not differ in dosage form and content of active ingredients from the corresponding originals. Bioequivalence studies allow us to make reasonable
conclusions about the quality of compared drugs based on a smaller volume of primary information and in a shorter period of time. They are carried out mainly on healthy volunteers.
Clinical trials of all phases are carried out in Russia. Most of the international clinical trials and studies of foreign medicines belong to the 3rd phase, and in the case of clinical trials of domestic medicines, a significant part of them are phase 4 studies.
In Russia, over the past ten years, a specialized clinical research market. It is well structured, highly qualified professionals work here - doctors-researchers, scientists, organizers, managers, etc., active enterprises that build their business on the organizational, service, analytical aspects of conducting clinical trials, among them - contract research organizations, medical centers statistics.
Between October 1998 and January 1, 2005, 1,840 clinical trials were submitted for approval. In 1998-1999 domestic companies made up an extremely small proportion of applicants, but since 2000 their role has increased noticeably: in 2001 there were 42%, in 2002 - already 63% of applicants, in 2003 - 45.5%. Among the foreign applicant countries, Switzerland, the USA, Belgium, and the UK take precedence.
The objects of study of clinical trials are medicines of both domestic and foreign origin, the scope of which affects almost all known areas of medicine. The largest number of drugs are used to treat cardiovascular diseases and cancer. This is followed by such areas as psychiatry and neurology, gastroenterology, and infectious diseases.
One of the trends in the development of the clinical trials sector in our country should be the rapid growth in the number of clinical trials on the bioequivalence of generic drugs. Obviously, this is fully consistent with the characteristics of the Russian pharmaceutical market: as is known, it is a market for generic drugs.
Conducting clinical trials in Russia is regulatedThe Constitution of the Russian Federation, which states that “... no one
may be subjected to medical, scientific and other experiments without voluntary consent.”
Some articles Federal Law “Fundamentals of the legislation of the Russian Federation on the protection of the health of citizens”(dated July 22, 1993, No. 5487-1) define the basics of conducting clinical trials. Thus, Article 43 states that medicines that are not approved for use, but are being reviewed in accordance with the established procedure, can be used in the interests of curing a patient only after receiving his voluntary written consent.
Federal Law “On Medicines” No. 86-FZ has a separate Chapter IX “Development, preclinical and clinical trials of medicinal products” (Articles 37-41). The procedure for making a decision on conducting a clinical trial of drugs, the legal basis for conducting clinical trials and issues of financing clinical trials, the procedure for conducting them, and the rights of patients participating in clinical trials are indicated here.
Clinical studies are conducted in accordance with Industry Standard OST 42-511-99 “Rules for conducting high-quality clinical trials in the Russian Federation”(approved by the Russian Ministry of Health on December 29, 1998) (Good Clinical Practice - GCP). The Rules for Conducting Quality Clinical Trials in the Russian Federation provide an ethical and scientific standard for the quality of design and conduct of human studies, and the documentation and presentation of their results. Compliance with these rules serves as a guarantee of the reliability of the results of clinical trials, safety, protection of the rights and health of subjects in accordance with the fundamental principles of the Declaration of Helsinki. The requirements of these Rules must be observed when conducting clinical trials of medicinal products, the results of which are planned to be submitted to licensing authorities.
GCP establishes requirements for the planning, conduct, documentation and control of clinical trials designed to ensure the protection of the rights, safety and health of persons participating in them, during which undesirable effects on human safety and health cannot be excluded, as well as to ensure the reliability and accuracy of the obtained data. during information research. The rules are mandatory for all participants in clinical trials of medicinal products on the territory of the Russian Federation.
In order to improve the methodological basis for conducting bioequivalence studies of medicines, which are the main type of medical and biological control of generic medicines, the Ministry of Health and Social Development of the Russian Federation approved methodological guidelines on August 10, 2004 “Conducting high-quality clinical studies of bioequivalence of drugs.”
According to regulatory documents, CIs are carried out in healthcare institutions accredited by the federal executive body, whose competence includes the implementation of state control and supervision in the field of circulation of medicines; it also compiles and publishes a list of healthcare institutions that have the right to conduct clinical trials of medicines.
Legal basis for conducting clinical trials of drugs constitute a decision of the federal executive body, whose competence includes the implementation of state control and supervision in the field of circulation of medicines, on conducting a clinical trial of a medicine and an agreement on its conduct. The decision to conduct a clinical trial of drugs is made by the Federal Service for Surveillance in Healthcare and Social Development of the Russian Federation in accordance with the Law “On Medicines” and on the basis of an application, a positive conclusion of the ethics committee under the federal body for quality control of medicines, a report and conclusion on preclinical research and instructions for medical use of the drug.
An Ethics Committee has been established under the federal body for drug quality control. The health care institution does not begin the study until the Ethics Committee has approved (in writing) the written informed consent form and other materials provided to the subject or his legal representative. The informed consent form and other materials may be revised during the course of the study if circumstances are discovered that could affect the consent of the subject. The new edition of the documentation listed above must be approved by the Ethics Committee, and the fact of its delivery to the subject must be documented.
For the first time in world practice, state control over the conduct of clinical trials and observance of the rights of experimental participants was developed and implemented in Prussia. On October 29, 1900, the Ministry of Health obliged university clinics to conduct clinical experiments subject to prior written consent from patients. In the 1930s The situation in the world has changed dramatically with regard to human rights. In concentration camps for prisoners of war in Germany and Japan, human experiments were carried out on such a large scale that over time, each concentration camp even developed its own “specialization” in medical experiments. Only in 1947 did the International Military Tribunal return to the problem of protecting the rights of people taking part in clinical research. During its work, the first international code was developed "Code of Rules for Conducting Experiments on Humans" the so-called Nuremberg Code.
In 1949, the International Code of Medical Ethics was adopted in London, proclaiming the thesis that “a doctor should act only in the interests of the patient, providing medical care that should improve the physical and mental condition of the patient,” and the Geneva Convention of the World Association of Physicians (1948 -1949), defined the duty of a doctor with the words: “caring for the health of my patient is my first task.”
A turning point in the development of the ethical framework for clinical trials was the adoption by the 18th General Assembly of the World Medical Association in Helsinki in June 1964. Declaration of Helsinki World Medical Association, which has absorbed all the world experience in the ethical content of biomedical research. Since then, the Declaration has been revised several times, most recently in Edinburgh (Scotland) in October 2000.
The Declaration of Helsinki states that biomedical research involving human subjects must conform to generally accepted scientific principles and be based on adequately conducted laboratory and animal experiments, as well as a sufficient knowledge of the scientific literature. They must be carried out by qualified personnel under the supervision of an experienced physician. In all cases, the doctor is responsible for the patient, but not the patient himself, despite the informed consent given by him.
In any research involving human subjects, each potential participant should be adequately informed of the purposes, methods, expected benefits of the research, and the risks and harms involved. People should be informed that they have the right to refrain from participating in the research and can withdraw their consent and refuse to continue the research at any time after the trial has begun. The physician must then obtain freely given written informed consent from the subject.
Another important document defining ethical standards for conducting clinical trials was "International Guide to the Ethics of Biomedical Research Involving Human Subjects" adopted by the Council of International Organizations of Medical Science (CIOMS) (Geneva, 1993), which provides guidance to researchers, funders, health officials and ethics committees on how to implement ethical standards in medical research, as well as ethical principles relevant to all persons, including patients participating in clinical trials.
The Declaration of Helsinki and the International Guidelines for the Ethics of Biomedical Research Involving Human Subjects show how fundamental ethical principles can be effectively applied in medical research practice throughout the world, taking into account the different circumstances of cultures, religions, traditions, social and economic conditions, laws, administrative systems and other situations that may occur in countries with limited resources.
On November 19, 1996, the Parliamentary Assembly of the Council of Europe adopted "Convention for the Protection of Human Rights and Human Dignity with regard to the Application of Biology and Medicine." The norms laid down in the Convention not only have the force of a moral appeal - each state that accedes to it undertakes the obligation to implement “its main provisions in national legislation.” According to the provisions of this Convention, the interests and welfare of the individual prevail over the interests of society and science. Any medical intervention, including intervention for research purposes, must be carried out in accordance with professional requirements and standards. The subject must receive in advance appropriate information about the purpose and nature of the intervention, as well as
its consequences and risks; his consent must be voluntary. Medical intervention in relation to a person who is unable to give consent may be carried out solely in his immediate interests. On January 25, 2005, the Additional Protocol to the Convention concerning biomedical research was adopted.
To ensure that the rights of research subjects are respected, the international community has now developed an effective system of public and government control over ensuring the rights and interests of research subjects and the ethics of clinical trials. One of the main links in the public control system is the activities of independent ethics committees(EC).
Ethics committees are today structures in the field of view of which scientific interests, medical facts and norms of morality and law intersect. Ethics committees carry out the functions of examination, consultation, recommendations, encouragement, evaluation, and guidance in moral and legal issues of CI. Ethics committees play a critical role in determining that research is safe, conducted in good faith, and that the rights of patients participating in it are respected; in other words, these committees provide assurance to the public that every clinical trial conducted meets ethical standards.
ECs must be independent of researchers and must not receive material benefits from the research being conducted. The researcher must obtain the advice, favorable review, or permission of the committee before commencing the work. The committee carries out further control, can amend the protocol and monitor the progress and results of the study. Ethics committees should have the power to prohibit research, terminate research, or simply refuse or terminate approval.
Basic principles for the activities of ethics committees When carrying out ethical examination, CIs are independence, competence, openness, pluralism, as well as objectivity, confidentiality, and collegiality.
ECs must be independent of the bodies making decisions on conducting clinical trials, including government bodies. An indispensable condition for the competence of the committee is the high qualifications and precise work of its protocol group (or
secretariat). The openness of the ethics committee's activities is ensured by the transparency of the principles of its work, regulations, etc. Standard operating procedures should be open to anyone who wishes to review them. The pluralism of the ethics committee is guaranteed by the diversity of professions, ages, genders, and religions of its members. The review process must take into account the rights of all study participants, in particular, not only patients, but also doctors. Confidentiality is required with respect to the materials of the trial and the persons participating in it.
An independent ethical committee is usually created under the auspices of national or local health departments, on the basis of medical institutions or other national, regional, local representative bodies - as a public association without forming a legal entity.
The main goals of the work of the ethics committee are the protection of the rights and interests of subjects and researchers; impartial ethical assessment of clinical and preclinical studies (trials); ensuring the conduct of high-quality clinical and preclinical studies (tests) in accordance with international standards; providing public confidence that all ethical principles will be guaranteed and observed.
To achieve these goals, the ethical committee must solve the following tasks: independently and objectively assess the safety and integrity of human rights in relation to the subjects, both at the planning stage and at the stage of conducting the study (test); assess the compliance of the study with humanistic and ethical standards, the feasibility of each study (test), compliance of the researchers, technical means, protocol (program) for the study, selection of study subjects, quality of randomization with the rules for conducting high-quality clinical trials; monitor compliance with quality standards for clinical trials to ensure the reliability and completeness of data.
Risk-benefit assessment is the most important ethical decision that the EC makes when reviewing research projects. To determine whether the risks are reasonable in relation to the benefits, a number of factors must be taken into account, and each case should be considered individually,
taking into account the characteristics of the subjects participating in the study (children, pregnant women, terminally ill patients).
To conduct an assessment of risks and expected benefits, the EC must ensure that:
The necessary data cannot be obtained without involving people in the research;
The study was rationally designed to minimize discomfort and invasive procedures for the subjects;
The research serves to obtain important results aimed at improving diagnosis and treatment or contributing to the generalization and systematization of data on diseases;
The research is based on the results of laboratory data and animal experiments, in-depth knowledge of the history of the problem, and the expected results will only confirm its validity;
The expected benefit from the study exceeds the potential risk, and the potential risk is minimal, i.e. no more than when performing conventional therapeutic and diagnostic procedures for this pathology;
The investigator has sufficient information about the foreseeability of any possible adverse consequences of the study;
Subjects and their legal representatives are provided with all information necessary to obtain their informed and voluntary consent.
Clinical trials must be carried out in accordance with the provisions of international and national legislative documents that guarantee protection of the rights of the subject.
The provisions contained in the Convention for the Protection of Human Rights protect the dignity and individual integrity of a person and guarantee to everyone, without exception, respect for the integrity of the person and other rights and fundamental freedoms in connection with the application of advances in biology and medicine, including in the field of transplantation, genetics, psychiatry and etc.
No human research can be conducted without simultaneously meeting all of the following conditions:
There are no alternative research methods that are comparable in their effectiveness;
The risk to which the subject may be exposed does not exceed the potential benefit from conducting this research;
The design of the proposed study has been approved by a competent authority following an independent review of the scientific merits of the study, including the importance of its purpose, and multilateral consideration of its ethical acceptability;
The person acting as a subject is informed of his rights and guarantees provided for by law;
Written informed consent to conduct the experiment was obtained, which can be freely withdrawn at any time.
The “Fundamentals of the Legislation of the Russian Federation on the Protection of Citizens’ Health” and the Federal Law “On Medicines” enshrine the provision that any biomedical research involving humans as an object must be carried out only after obtaining the written consent of the citizen. A person cannot be forced to participate in biomedical research.
Upon receipt of consent For biomedical research, a citizen must be provided with the following information:
1) about the medicinal product and the nature of its clinical trials;
2) about the expected effectiveness, the safety of the drug, the degree of risk for the patient;
3) about the patient’s actions in the event of unforeseen effects of the drug on his health;
4) about the conditions of the patient’s health insurance.
The patient has the right to refuse to participate in clinical trials at any stage.
Information about the study should be communicated to the patient in an accessible and understandable form. Before obtaining informed consent, the investigator or investigator must give the subject or his representative sufficient time to decide whether to participate in the study and provide the opportunity to obtain detailed information about the trial.
Informed consent (informed patient consent) ensures that future subjects understand the nature of the study and can make an informed and voluntary decision
about your participation or non-participation. This guarantee protects all parties: both the subject, whose autonomy is respected, and the researcher, who otherwise runs afoul of the law. Informed consent is one of the main ethical requirements for research involving human participants. It reflects the fundamental principle of respect for individuals. Elements of informed consent include full disclosure, adequate understanding, and voluntary choice. Various groups of the population may be involved in medical research, but it is prohibited to conduct clinical trials of drugs on:
1) minors without parents;
2) pregnant women, except in cases where clinical trials of medicinal products intended for pregnant women are conducted and when the risk of harm to the pregnant woman and the fetus is completely excluded;
3) persons serving sentences in places of deprivation of liberty, as well as those in custody in pre-trial detention centers without their written informed consent.
Clinical trials of medicinal products on minors are permitted only when the drug being studied is intended exclusively for the treatment of childhood diseases or when the purpose of the clinical trial is to obtain data on the best dosage of the drug for the treatment of minors. In the latter case, clinical trials on children should be preceded by similar trials on adults. In Art. 43 of the Fundamentals of the Legislation of the Russian Federation “on the protection of the health of citizens” it is noted: “Diagnostic, treatment methods and medications that are not approved for use, but are under consideration in accordance with the established procedure, can be used to treat persons under the age of 15 years only if there is an immediate threat to their life and with the written consent of their legal representatives." Information about the study should be communicated to children in a language that is age-appropriate for them to understand. Signed informed consent can be obtained from children who have reached the appropriate age (from 14 years of age, as determined by law and ethical committees).
Clinical trials of drugs intended for the treatment of mental illness are permitted on persons with mental illness and those recognized as legally incompetent.
established by the Law of the Russian Federation No. 3185-1 of July 2, 1992 “On psychiatric care and guarantees of the rights of citizens during its provision.” Clinical trials of medicinal products in this case are carried out with the written consent of the legal representatives of these persons.
Chapter 3. CLINICAL STUDIES OF DRUGS
The emergence of new drugs is preceded by a long cycle of studies, the task of which is to prove the effectiveness and safety of the new drug. The principles of preclinical research in laboratory animals were well established, but in the 1930s it became clear that the results obtained in animal experiments could not be directly transferred to humans.
The first clinical studies in humans were conducted in the early 1930s (1931 - the first randomized blind trial of sanocrysin** 3, 1933 - the first placebo-controlled study in patients with angina pectoris). Currently, several hundred thousand clinical studies have been conducted worldwide (30,000-40,000 per year). The appearance of each new drug is preceded by an average of 80 different studies involving more than 5,000 patients. This significantly lengthens the development period for new drugs (on average 14.9 years) and requires significant costs: manufacturing companies spend an average of $900 million on clinical trials alone. However, only clinical trials guarantee the receipt of accurate and reliable information about the safety and effectiveness of a new drug. drug.
According to the international guidelines for Good Clinical Practice (international standard for clinical research: ICH/GCP), under clinical trial understand “a study of the safety and/or effectiveness of an investigational drug in humans, aimed at identifying or confirming the clinical, desired pharmacodynamic properties of the investigational drug and/or conducted to identify its side effects and/or to study its absorption, distribution, biotransformation and excretion” .
Purpose of the clinical trial- obtaining reliable data on the effectiveness and safety of the drug without exposing
in this case, patients (research subjects) are exposed to unreasonable risks. More specifically, the study may aim to study the pharmacological effect of the drug on humans, establish therapeutic (therapeutic) effectiveness or confirm effectiveness in comparison with other drugs, as well as determine the therapeutic use - the niche that this drug can occupy in modern pharmacotherapy. In addition, research can be a stage in preparing a drug for registration, help promote an already registered drug on the market, or be a tool for solving scientific problems.
3.1. STANDARDS FOR CLINICAL TRIALS
Before the advent of uniform standards for clinical trials, patients receiving new drugs were often exposed to serious risks associated with taking insufficiently effective and dangerous drugs. For example, at the beginning of the twentieth century. in a number of countries, heroin was used as a cough treatment; in 1937 in the USA, several dozen children died after taking paracetamol syrup, which included toxic ethylene glycol *; and in the 1960s in Germany and the UK, approximately 10,000 children were born with severe limb abnormalities to women who took thalidomide* during pregnancy. Incorrect planning of studies, errors in the analysis of results and outright falsifications have caused a number of other humanitarian disasters, which raised the question of legislative protection of the interests of patients participating in studies and potential consumers of drugs.
Today, the potential risk of prescribing new drugs is significantly lower, since government agencies that give their approval for their use have the opportunity to evaluate the results of using a new drug in thousands of patients during clinical trials performed according to a single standard.
Currently, all clinical studies are carried out according to a single international standard, called GCP. , which was developed by the Drug Control Administration
supplies and food products from the US government, WHO and the European Union in the 1980s and 1990s. The GCP standard regulates the planning and conduct of clinical trials, and also provides for multi-stage monitoring of patient safety and the accuracy of the data obtained.
The GCP standard takes into account the ethical requirements for scientific research involving human subjects, formulated by Declaration of Helsinki of the World Medical Association"Recommendations for Clinicians Conducting Biomedical Research Involving Human Subjects." In particular, participation in clinical trials can only be voluntary; patients should not receive monetary compensation during the research. By signing his consent to become a study participant, the patient receives accurate and detailed information about the possible risk to his health. In addition, the patient can stop participating in the study at any time without giving any reason.
Clinical pharmacology, which studies the pharmacokinetics and pharmacodynamics of drugs directly in a sick person, was of great importance in the creation of GCP standards and the entire modern concept of clinical trials of drugs.
The provisions of the international standard ICH GCP are reflected in Federal Law “On Circulation of Medicines”(No. 61-FZ dated April 12, 2010) and State standard "Good clinical practice"(GOST R 52379-2005), according to which clinical trials of drugs are carried out in our country. Thus, there is a legal basis for the mutual recognition of the results of clinical trials between different countries, as well as for the conduct of large international clinical trials.
3.2. PLANNING AND CONDUCTING CLINICAL STUDIES
Planning a clinical trial involves several stages.
Definition of the research question. For example, does drug X significantly reduce blood pressure in patients with hypertension, or does drug X actually lower blood pressure more effectively than drug Y? A study typically has one main study and several additional study arms.
questions, for example: can drug Z reduce mortality in patients with hypertension (main question), how does drug Z affect the frequency of hospitalizations, what is the proportion of patients with moderate hypertension in whom drug Z is able to reliably control blood pressure levels (additional questions). The research question reflects the assumption from which the researchers start (research hypothesis); in our example, the hypothesis is that drug Z, having the ability to lower blood pressure, can reduce the risk of hypertension-related complications and diseases and, therefore, can reduce the incidence of deaths.
Selecting a study design. The study may include several comparison groups (drug A and placebo or drug A and drug B). Studies that do not have a comparison group do not provide reliable information about the effects of drugs, and at present such studies are practically not carried out.
Determining the sample size. The authors of the protocol must foresee exactly how many patients will be needed to prove the initial hypothesis (the sample size is calculated mathematically based on the laws of statistics). The study can include from several dozen (in the case when the effect of the drug is significantly pronounced) to 30,000-50,000 patients (if the effect of the drug is less pronounced).
Determining the duration of the study. The duration of the study depends on the time of onset of the effect. For example, bronchodilators improve the condition of patients with bronchial asthma within a few minutes after taking them, but it is possible to register the positive effect of inhaled glucocorticoids in these patients only after several weeks. In addition, some studies require observation of relatively rare events: if the study drug is expected to reduce the number of exacerbations of the disease, then long-term follow-up is necessary to confirm this effect. In modern studies, observation periods range from several hours to 5-7 years.
Selection of patient population. To include patients with certain characteristics into the study, developers create clear criteria. They include age, gender, duration and severity of the disease, the nature of the previous
treatment, concomitant diseases that may affect the assessment of the effect of the drug. Inclusion criteria should ensure homogeneity of patients. For example, if a hypertension trial were to simultaneously enroll patients with mild (borderline) hypertension and patients with very high blood pressure, the study drug would affect these patients differently, making it difficult to obtain reliable results. In addition, studies usually do not include pregnant women and people with severe diseases that negatively affect the general condition and prognosis of the patient.
Methods for assessing the effectiveness of treatment. Developers must select indicators of the effectiveness of the drug; in our example, it is necessary to clarify how exactly the hypotensive effect will be assessed - by a single measurement of blood pressure; by calculating the average daily blood pressure; the effectiveness of treatment will be assessed by the effect on the patient’s quality of life or by the ability of the drug to prevent the manifestations of complications of hypertension.
Safety assessment methods. Measures should be in place to assess the safety of treatment and methods for recording ADRs of investigational drugs.
The planning stage ends with the writing of a protocol - the main document that provides for the conduct of the study and all research procedures. Thus, research protocol“describes the objectives, methodology, statistical aspects, and organization of the study.” The protocol is provided for review to government regulatory authorities and an independent ethical committee, without whose approval the study cannot begin. Internal (monitoring) and external (audit) control over the study evaluates, first of all, the compliance of the researchers’ actions with the procedure described in the protocol.
Inclusion of patients in the study- purely voluntary. A mandatory condition for inclusion is that the patient is familiarized with the possible risks and benefits that he can derive from participating in the study, as well as signing informed consent. ICH GCP rules do not allow the use of financial incentives to attract patients to participate in a study (an exception is made for healthy volunteers recruited to study the pharmacokinetics or bioequivalence of drugs). The patient must meet the inclusion/exclusion criteria. Usually
Pregnant women, nursing mothers, patients in whom the pharmacokinetics of the study drug may be altered, and patients with alcoholism or drug addiction are not allowed to participate in studies. It is unacceptable to include incapacitated patients into the study without the consent of caregivers, military personnel, prisoners, persons with an allergy to the study drug, or patients who are simultaneously participating in another study. The patient has the right to stop participating in the study at any time without giving reasons.
Study design. Studies in which all patients receive the same treatment are currently practically non-existent due to the low evidence of the results obtained. The most common comparative study is parallel group (intervention and control group). A placebo (placebo-controlled trial) or another active drug can be used as a control.
Studies with comparative designs require randomization- random distribution of participants into experimental and control groups, which allows to minimize systematic error and bias. The researcher can, in principle, gain access to information about which drug the patient is receiving (this may be necessary if serious adverse reactions occur), but in this case the patient must be excluded from the study.
Individual registration card. An individual registration card is defined as “a printed, optical, or electronic document created to record all protocol-required information about each study subject.” Based on the individual registration card, a research database is created for statistical processing of the results.
3.3. PHASES OF CLINICAL DRUG TRIALS
Both the manufacturer and the public are interested in obtaining the most accurate and complete information about the clinical pharmacology, therapeutic efficacy and safety of a new drug during pre-registration studies. Preparation
registration dossier is impossible without answers to these questions. Because of this, the registration of a new drug is preceded by several dozen different studies, and both the number of studies and the number of their participants increases every year, and the total research cycle of a new drug usually exceeds 10 years. Thus, the development of new drugs is possible only in large pharmaceutical companies, and the total cost of a research project on average exceeds $900 million.
The first, preclinical studies begin soon after the synthesis of a new, potentially effective molecule. Their essence is to test the hypothesis about the expected pharmacological action of a new compound. At the same time, the toxicity of the compound, its oncogenic and teratogenic effects are being studied. All these studies are performed on laboratory animals, and their total duration is 5-6 years. As a result of this work, approximately 250 are selected from 5-10 thousand new compounds.
Clinical trials themselves are conventionally divided into four periods or phases.
Phase I clinical trials, usually carried out on 28-30 healthy volunteers. The purpose of this stage is to obtain information about the tolerability, pharmacokinetics and pharmacodynamics of the new drug, clarify the dosage regimen and obtain data on the safety of the drug. Studying the therapeutic effect of the drug in this phase is not necessary, since a number of clinically important properties of the new drug are usually not observed in healthy volunteers.
Phase I studies begin with a study of the safety and pharmacokinetics of a single dose, the selection of which is based on data obtained from biological models. In the future, the pharmacokinetics of the drug with repeated administration, excretion and metabolism of the new drug (the order of kinetic processes), its distribution in fluids and body tissues, and pharmacodynamics are studied. Typically, all these studies are carried out for different doses, dosage forms and routes of administration. During phase I studies, it is also possible to evaluate the effect of other drugs, the functional state of the body, food intake, etc. on the pharmacokinetics and pharmacodynamics of a new drug.
An important goal of phase I clinical trials is to identify potential toxicities and ADRs, but these studies are of short duration and are carried out in a limited number of participants, therefore, during this phase it is possible to identify only the most
frequent and severe adverse events associated with the use of new drugs.
In some cases (oncology drugs, drugs for the treatment of HIV infection), phase I studies can be carried out in patients. This makes it possible to speed up the creation of a new drug and not expose volunteers to unreasonable risks, although this approach can be considered more of an exception.
Phase I studies allow:
Assess the tolerability and safety of a new drug;
In some cases, get an idea of its pharmacokinetics (in healthy people, which naturally has limited significance);
Determine the main pharmacokinetic constants (Cmax,
C1);
Compare the pharmacokinetics of a new drug using different dosage forms, routes and methods of administration.
Phase II studies- first studies in patients. The volume of these studies is significantly larger than in phase I: 100-200 patients (sometimes up to 500). In phase II, the effectiveness and safety of the new drug, as well as the dosage range for treating patients, are clarified. These studies provide information mainly on the pharmacodynamics of the new drug. Comparative design and inclusion of a control group are considered mandatory conditions for conducting phase II studies (which is not typical for phase I studies).
Phase III studies are planned for a large number of patients (up to 10,000 people or more), and the conditions for their implementation are as close as possible to the usual conditions for the treatment of certain diseases. Studies in this phase (usually several parallel or sequential studies) are large (full-scale), randomized and comparative. The subject of study is not only the pharmacodynamics of the new drug, but also its clinical effectiveness 1 .
1 For example, the goal of a study of a new antihypertensive drug in phases I-II is to prove its ability to lower blood pressure, and in a phase III study the goal is to study the effect of the drug on hypertension. In the latter case, along with a decrease in blood pressure, other points for assessing the effect appear, in particular, a decrease in mortality from cardiovascular diseases, prevention of complications of hypertension, improvement in the quality of life of patients, etc.
In phase III studies, the drug is compared in terms of effectiveness and safety with a placebo (placebo-controlled study) or/and with another marker drug (a drug commonly used in a given clinical situation and with well-known medicinal properties).
Submission by the developer of an application for drug registration does not mean the completion of research. Phase III studies performed before filing an application are called phase Ia studies, and those performed after filing an application are called phase III studies. The latter are carried out to obtain more complete information about the clinical and pharmacoeconomic effectiveness of drugs. Such studies may expand the indications for prescribing a new drug. Additional studies may be initiated by government agencies responsible for the registration process if the results of previous studies do not allow an unambiguous statement about the properties and safety of the new drug.
The results of phase III studies become decisive when deciding to register a new drug. This decision may be made if the drug:
More effective than already known drugs of similar action;
Has effects that are not characteristic of existing drugs;
Has a more advantageous dosage form;
More beneficial in pharmacoeconomic terms or allows the use of simpler treatment methods;
It has advantages when used together with other drugs;
Has an easier way to use.
Phase IV studies. Competition with new drugs forces research to continue even after registration of a new drug (post-marketing studies) to confirm the effectiveness of the drug and its place in pharmacotherapy. In addition, phase IV studies make it possible to answer some questions that arise during the use of drugs (the optimal duration of treatment, the advantages and disadvantages of a new drug in comparison with others, including newer drugs, features of prescription in the elderly, children, long-term effects of treatment, new indications, etc.).
Sometimes phase IV studies are carried out many years after drug registration. An example of such deferrals of more than 60 years
Clinical studies of all phases are carried out in 2 centers officially certified by state control authorities (medical centers, hospitals, clinics), which have the appropriate scientific and diagnostic equipment and the ability to provide qualified medical care to patients with ADRs.
Bioequivalence studies. Most drugs on the pharmaceutical market are reproduced (generic) drugs. The pharmacological action and clinical effectiveness of the drugs included in these drugs, as a rule, have been quite well studied. However, the effectiveness of generics can vary significantly.
Registration of generic drugs can be simplified (in terms of time and scope of research). Bioequivalence studies allow us to make a strictly substantiated conclusion about the quality of these products. In these studies, the generic drug is compared with the original drug in terms of bioavailability (the proportion of the drug reaching the systemic circulation and the rate at which this process occurs are compared). If two drugs have the same bioavailability, they are bioequivalent. It is assumed that bioequivalent drugs have the same effectiveness and safety 3 .
Bioequivalence is studied on a small number of healthy volunteers (20-30), using standard procedures for studying pharmacokinetics (construction of a pharmacokinetic curve, studies of AUC, Tmax, Cmax values).
max max
1 Proposed into clinical practice about 100 years ago, these drugs at one time did not undergo the process of registration and clinical trials, which required their extensive research more than 60 years later. The modern system for registering new drugs appeared in the 60s of the 20th century, therefore, about 30-40% of drugs used today have not been convincingly studied. Their place in pharmacotherapy may be a matter of debate. In the English-language literature, the term “orphan drugs” is used for these drugs, since it is rarely possible to find sources of funding for research on such drugs.
2 In our country - the Ministry of Health and Social Development of the Russian Federation.
3 However, it cannot be stated that two pharmaceutically equivalent drugs (with the same efficacy and safety) always have the same pharmacokinetics and comparable bioavailability.
3.4. ETHICAL ASPECTS OF CLINICAL
RESEARCH
The most important principle of medical ethics was formulated almost 2500 years ago. The Hippocratic Oath states: “I undertake to do all this according to my ability and knowledge for the benefit of the patient and to abstain from everything that can cause him harm.” The requirements of medical deontology acquire particular importance when conducting clinical trials of drugs because they are conducted on people and affect human rights to health and life. Consequently, medico-legal and medico-deontological problems are of great importance in clinical pharmacology.
When conducting clinical trials of drugs (both new and already studied, but used for new indications), one should be guided primarily by the interests of the patient. Permission to conduct clinical trials of drugs is accepted by the competent authorities (in the Russian Federation - the Ministry of Health and Social Development of Russia) after a detailed study of the totality of data obtained during the preclinical study of the drug. However, regardless of government approval, the study must also receive approval from an ethics committee.
Ethical review of clinical research is carried out in accordance with the principles of the Declaration of Helsinki of the World Medical Association “Recommendations for physicians engaged in biomedical research involving human subjects” (first adopted by the 18th World Medical Assembly in Helsinki in 1964 and subsequently amended and revised several times).
The Declaration of Helsinki states that the purpose of biomedical research in humans should be to improve diagnostic, therapeutic and preventive procedures, as well as to elucidate the etiology and pathogenesis of diseases. The World Medical Assembly has prepared recommendations for physicians when conducting clinical trials.
The requirements of the Declaration of Helsinki were taken into account in the Federal Law of the Russian Federation “On the Circulation of Medicines”. In particular, the following is confirmed by law.
Participation of patients in clinical trials of drugs can only be voluntary.
The patient gives written consent to participate in clinical trials of drugs.
The patient must be informed about the nature of the study and the possible risk to his health.
The patient has the right to refuse to participate in clinical trials of drugs at any stage.
According to ethical requirements, clinical trials of drugs in relation to minors (except for those cases when the drug being studied is intended exclusively for the treatment of childhood diseases) and pregnant women are prohibited. It is prohibited to conduct clinical trials of drugs in minors without parents, incompetent persons, prisoners, military personnel, etc. All participants in clinical trials must be insured.
The issues of ethical review of clinical trials in our country are dealt with by the ethics committee of the Ministry of Health and Social Development of Russia, as well as local ethics committees at medical and scientific medical institutions. The Ethics Committee is guided by the basic international principles of conducting clinical research, as well as the current legislation and regulations of the Russian Federation.
3.5. PROCEDURE FOR REGISTRATION OF NEW MEDICINES
According to the Federal Law “On the Circulation of Medicines” (No. 61-FZ dated April 12, 2010), “Medicines can be produced, sold and used on the territory of the Russian Federation if they are registered by the federal body for drug quality control.” The following are subject to state registration:
New drugs;
New combinations of previously registered drugs;
Medicines registered earlier, but produced in other dosage forms or in a new dosage;
Generic drugs.
The state registration of drugs is carried out by the Ministry of Health and Social Development of Russia, it also approves the instructions for the use of drugs, and the registered drug is entered into the state register.
Clinical pharmacology and pharmacotherapy: textbook. - 3rd ed., revised. and additional / ed. V. G. Kukesa, A. K. Starodubtseva. - 2012. - 840 p.: ill.
1. Clinical trials of medicinal products for medical use, including international multicenter, multicenter, post-registration, are conducted in one or more medical organizations in accordance with the rules of good clinical practice approved by the authorized federal executive body, respectively, for the following purposes:
1) establishing the safety of medicinal products for healthy volunteers and (or) their tolerability by healthy volunteers, with the exception of such studies of medicinal products produced outside the Russian Federation;
3) establishing the safety of the drug and its effectiveness for patients with a certain disease, the preventive effectiveness of immunobiological drugs for healthy volunteers;
4) studying the possibility of expanding indications for medical use and identifying previously unknown side effects of registered drugs.
2. In relation to generic medicinal products for medical use, bioequivalence and (or) therapeutic equivalence studies are carried out in the manner established by the authorized federal executive body.
3. The organization of clinical trials of a medicinal product for medical use has the right to be carried out by:
1) the developer of the medicinal product or a person authorized by him;
2) educational organizations of higher education, organizations of additional professional education;
(see text in the previous edition)
3) research organizations.
4. Clinical trials of a medicinal product for medical use are carried out on the basis of a permit to conduct a clinical trial of a medicinal product issued by the authorized federal executive body. The authorized federal executive body maintains a register of issued permits to conduct clinical trials of a medicinal product, containing an indication of their purpose or purposes, in the manner established by this body.
(see text in the previous edition)
(see text in the previous edition)
6. Legal entities of any organizational and legal form may be involved in organizing clinical trials of a medicinal product for medical use by the developer of the medicinal product, provided that these studies comply with the requirements of this Federal Law.
7. Clinical trials of medicinal products for medical use are carried out in medical organizations accredited by the authorized federal executive body in the manner established by the Government of the Russian Federation.
8. The list of medical organizations that have the right to conduct clinical trials of medicinal products for medical use and the register of issued permits to conduct clinical trials of medicinal products are published and posted by the authorized federal executive body in the manner established by it on its official website on the Internet.

In 2018, the Nativity Fast will begin on November 28. During this period, Orthodox believers prepare to celebrate Christmas...

Starting a family is the dream of most women. They want to have a loving husband and a bunch of kids. But it's not always a relationship...

This article contains: the most powerful prayer for divorce - information taken from all over the world, the electronic network and...

Information site about icons, prayers, Orthodox traditions. Prayer for scandals and quarrels in the family, with husband, with children...

What would New Year be without champagne, tangerines, Olivier, aspic and everyone’s favorite “Herring under a fur coat”. With the last one...

Let's prepare the necessary ingredients for the cookies. The first thing to do is put the water to boil. We need...

Is it possible to register an employee for the position of financial director - chief accountant? The chief accountant claims...

The head of a small business can easily manage the budget independently. CHECKED! If you manage...

The creation of new projects involves a preliminary economic justification for their feasibility, subsequent...
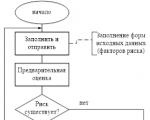
Reporting is generated by the RM, is agreed upon (approved) by the Risk Committee under the Management Board and transmitted to...

At the edge of a large, very large meadow, on a long emerald blade of grass lived a tiny Ladybug. God's little...

Nowadays, it is quite common for people to turn to the stars. With the help of a horoscope a person can find out...

Business or friendly. If you were pursuing a stranger, it means your level of trust in the female sex...

according to Freud's dream book If you dreamed about how you were fishing, it means that in real life you can hardly switch off...

Starting a family is the dream of most women. They want to have a loving husband and a bunch of kids. But it's not always...

This article contains: the most powerful prayer for divorce - information taken from all over the world, electronic...