A interpretação mais completa do sono laranja
Suco de laranja. O significado simbólico do suco de laranja nos livros de sonhos é prazer e tentação. Muitas vezes nós...
Cerca de 40% de todas as erupções hemorrágicas estão associadas à púrpura trombocitopênica. Sua prevalência varia de 1 a 13 pessoas por 100 mil habitantes, dependendo da região.
Entre todas as diáteses hemorrágicas, a púrpura trombocitopênica ocupa o primeiro lugar em prevalência entre crianças pré-escolares; em adultos, a síndrome é menos comum e afeta principalmente mulheres.
Púrpura trombocitopênica (Doença de Werlhof, PTI, trombocitopenia imune primária) é uma doença caracterizada por um nível reduzido de plaquetas no sangue ().
A vida útil das plaquetas varia de 7 a 10 dias.
Na púrpura trombocitopênica, o sistema imunológico suprime suas próprias células sanguíneas e elas morrem em poucas horas. A consequência disso é o aumento do tempo de sangramento.
A púrpura trombocitopênica é dividida nas formas aguda, recorrente e crônica da doença.
Dependendo da causa da doença, a púrpura trombocitopênica é dividida em formas:
As razões para o desenvolvimento da púrpura trombocítica ainda não estão totalmente estabelecidas.
A púrpura trombocitopênica também pode ocorrer devido a:
A púrpura trombocitopênica pode ocorrer em crianças e adultos de qualquer idade, mas existem fatores que aumentam o risco de desenvolver esta condição:

Na doença de Werlhof, os principais sintomas são hemorragias.

Foto de púrpura trombocitopênica idiopática
A doença de Werlhof tem três estágios em seu curso:
Os primeiros sinais de trombocitopenia aparecem quando o nível de plaquetas no sangue diminui abaixo de 50*109/l, isto ocorre aproximadamente 2-3 semanas após a exposição do fator que causou a doença.
 Sintomas agudos
Sintomas agudos A doença começa repentinamente: aparecem hemorragias na pele e nas mucosas (ver foto acima), começa o sangramento, piora o estado geral, a pele fica pálida e a pressão arterial cai.
A temperatura corporal pode chegar a 38 graus. Os gânglios linfáticos ficam inflamados e doloridos.

Petéquias na superfície lateral da língua.

Sinais de doença crônica
O principal sintoma da doença é a erupção cutânea. Eles aparecem na pele e nas mucosas, são dolorosos e variam em tamanho.
As erupções cutâneas podem ser de vários tipos:
A cor das erupções cutâneas recentes é roxa. As erupções cutâneas desbotadas são amarelas ou verdes. A erupção na pele pode ser úmida ou seca.
Em caso de erupções cutâneas úmidas, pode ocorrer sangramento, especialmente à noite.
Localização comum: no tórax, abdômen, extremidades superiores e inferiores, raramente na face e pescoço. Ao mesmo tempo, aparece uma erupção cutânea nas membranas mucosas.
Alguns locais são inacessíveis para inspeção sem equipamento especial: o tímpano, a membrana serosa do cérebro e outros órgãos.
Um sintoma importante da doença é o sangramento de intensidade variável. O sangramento mais comumente observado:
O sangramento ocorre simultaneamente com erupções cutâneas ou depois.
A temperatura corporal na púrpura trombocitopênica crônica não é elevada, às vezes há batimentos cardíacos acelerados e, em crianças, os gânglios linfáticos ficam aumentados e doloridos.

Esta forma da doença é a mais perigosa de todas.
Caracterizada por início agudo, espontâneo e curso maligno.
Devido à formação de coágulos sanguíneos hialinos, o fornecimento de sangue a vários órgãos é interrompido.
A púrpura trombocitopênica idiopática (PTI) do tipo trombótico é caracterizada pelos seguintes sintomas:
Para fazer o diagnóstico da doença de Werlhof, um paciente é entrevistado e examinado. Métodos laboratoriais para exame de sangue, urina e medula espinhal são utilizados para confirmar a presença da doença.
Nos casos crônicos, os parâmetros hematológicos podem estar dentro dos limites da normalidade.
Durante a entrevista, um hematologista (médico especialista em doenças do sangue) descobre a presença de fatores no histórico médico que contribuem para o desenvolvimento da púrpura trombocitopênica: infecção viral, medicamentos, vacinação, exposição à radiação, etc.
O exame do paciente revela um sinal característico desta doença - erupção cutânea hemorrágica na pele e nas mucosas. O médico também pode realizar uma série de exames que permitem detectar hemorragias na pele:
Um exame de sangue geral revela desvios no nível de hemoglobina (observados com perda significativa de sangue) e plaquetas. mostra a taxa de coagulação sanguínea, a presença de anticorpos antiplaquetários, redução ou ausência de retração do coágulo. Os glóbulos vermelhos são detectados.
Em casos graves, é realizada uma biópsia da medula óssea vermelha. Ao examinar uma amostra de biópsia, é revelado um conteúdo normal ou aumentado de megacariócitos e a presença de suas formas imaturas.
No caso de sintomas clínicos característicos, o tratamento pode ser iniciado imediatamente, sem aguardar o resultado dos exames laboratoriais.
Se a púrpura trombocitopênica não causar complicações, não houver sangramento significativo, a contagem de plaquetas no sangue não for inferior a 50 * 109 / l, as táticas médicas consistem em observação - nenhum tratamento é necessário.
Quando o nível de plaquetas diminui para 30-50*109/l, o tratamento é necessário para pacientes com risco de sangramento (hipertensão arterial).
Se o nível de plaquetas cair abaixo de 30*109/l, é necessária hospitalização urgente.

A terapia conservadora inclui o uso de medicamentos que suprimem processos autoimunes e reduzem a permeabilidade vascular:
Para estancar o sangramento externo, utiliza-se uma esponja hemostática; em caso de sangramento interno, são administrados medicamentos para estancar o sangramento.
Em casos graves, é utilizada a plasmaférese - transfusão de hemocomponentes e plaquetas.
Em alguns casos, a púrpura trombocitopênica idiopática não responde à terapia conservadora, sendo então realizada a cirurgia de remoção - esplenectomia.
Uma melhora significativa é observada imediatamente, mas há risco de complicações pós-operatórias e a resistência do organismo a doenças infecciosas é significativamente reduzida.
A esplenectomia é realizada para diversas indicações:
Para tratar a púrpura trombocítica, além de medicamentos, são utilizadas plantas medicinais com propriedades hemostáticas. Entre eles:
É importante que os alimentos consumidos sejam ligeiramente quentes ou frios. Legumes e frutas frescas são úteis, mas você precisa ter certeza de que não causam uma reação alérgica.
Entrada:
Em adultos, a púrpura trombocitopênica termina em recuperação completa em 75% dos casos, em crianças - em 90% dos casos. A ocorrência de complicações graves só é possível no período agudo da doença.
A probabilidade de morte na variante trombótica da púrpura trombocitopênica depende da extensão da lesão e do grau de dano ao cérebro, sistema cardiovascular, rins e outros órgãos.
Pacientes com histórico dessa patologia necessitam de supervisão médica constante, exclusão de medicamentos que afetam negativamente a coagulação sanguínea e revisão do estilo de vida e nutrição.
As medidas preventivas para a púrpura trombocitopênica visam prevenir exacerbações. Eles envolvem a manutenção dos níveis de plaquetas e hemoglobina no sangue e incluem:
Após a alta hospitalar, o paciente é cadastrado no dispensário de seu local de residência - é observado por um médico há pelo menos 2 anos.
No entanto, uma grande parte da responsabilidade pela saúde recai sobre os ombros do paciente ou dos seus pais em caso de doença de uma criança.
Dado que a púrpura trombocitopênica é comum em crianças, é muito importante educar toda a família sobre como prevenir esta doença.
Em adultos, este é um distúrbio que pode causar grandes hematomas e sangramentos rapidamente. Normalmente, a perda de sangue ocorre devido a uma queda extrema no nível de plaquetas – células que ajudam a coagular o sangue.
Em algumas fontes, a patologia em questão também é chamada de trombocitopenia imune. Afeta adultos e crianças, mas em pacientes jovens essa condição geralmente se desenvolve no contexto de uma infecção viral e desaparece por conta própria, sem necessidade de tratamento especial. Nos adultos, a doença na maioria dos casos é prolongada.
Se você não tiver sinais de sangramento e sua contagem de plaquetas, embora baixa, for geralmente satisfatória, seu médico provavelmente não prescreverá tratamento. Em casos raros, contudo, a contagem de plaquetas cai para um mínimo crítico, causando hemorragias internas potencialmente fatais. Para tratar tais complicações, é necessária intervenção médica imediata.
Em adultos (foto acima) nem sempre é acompanhada de manifestações externas de patologia. Se ocorrerem sintomas característicos, os seguintes sinais da doença podem ser observados:
A púrpura trombocitopênica em adultos pode ser uma preocupação séria; nesse caso, é melhor marcar uma consulta com um especialista qualificado com antecedência.

Se você estiver sangrando, chame uma ambulância. O sangramento característico desta doença não pode ser controlado com remédios caseiros convencionais ou métodos típicos de interrupção do fluxo sanguíneo. Assim, por exemplo, a pressão física na parte correspondente do corpo não o ajudará. Os especialistas recomendam procurar ajuda médica o mais rápido possível.
Às vezes, a púrpura em adultos se desenvolve porque o sistema imunológico do corpo ataca e destrói as plaquetas por engano. O termo "idiopático" significa "desenvolvido a partir de uma causa desconhecida"; A trombocitopenia é tão caracterizada nos casos em que os médicos não conseguem determinar o que exatamente provocou uma reação inadequada do sistema imunológico.
Em crianças, geralmente é mais fácil estabelecer os pré-requisitos para a patologia, uma vez que esse distúrbio geralmente ocorre após uma doença infecciosa (caxumba ou gripe). Presume-se que é a infecção que perturba o funcionamento normal do sistema imunológico.
Se o diagnóstico inicial for púrpura trombocitopênica em adultos, as manifestações clínicas geralmente estão associadas a um distúrbio imunológico. Os anticorpos se ligam às plaquetas, marcando-as como células a serem destruídas. O baço, projetado para combater infecções externas, reconhece anticorpos e remove plaquetas do corpo. Como resultado desta identidade equivocada, há muito menos plaquetas circulando no sangue do que o normal.

A contagem normal de plaquetas varia de 150 a 450 mil células por microlitro de sangue circulante. Em pessoas com diagnóstico de púrpura trombocitopênica (adultos), os exames revelam apenas 20 mil das células necessárias. Como as plaquetas ajudam a coagular o sangue, uma diminuição no seu número leva a um risco aumentado de sangramento e hemorragia interna. Um nível de 10 mil células é fatal. Com uma diminuição tão significativa no número de plaquetas, a hemorragia interna pode começar mesmo sem qualquer trauma.
A púrpura trombocitopênica em adultos (a foto usada no início do artigo demonstra as manifestações externas da patologia) é detectada em qualquer idade, mas os cientistas identificaram dois sinais da doença que aumentam a probabilidade de desenvolvimento. Esse:
Você deve tomar cuidado com o risco de complicações. O mais grave deles é aquele que pode ser fatal.
Embora a púrpura trombocitopênica em adultos seja diagnosticada mesmo em gestantes, na maioria dos casos essa patologia materna não afeta a saúde do recém-nascido. Os especialistas, porém, recomendam verificar o nível de plaquetas no sangue do bebê imediatamente após o nascimento.

Se você estiver grávida e os resultados dos exames indicarem uma diminuição nos níveis de plaquetas, você corre risco, pois a probabilidade de sangramento pós-parto grave é muito alta. Nesses casos, o ginecologista observador ou os médicos da maternidade devem discutir com você os métodos de tratamento disponíveis que são seguros para a criança.
Para que o médico faça um diagnóstico preciso de “púrpura trombocitopênica” (em adultos), o diagnóstico deve ser feito de acordo com o princípio diferencial. Você deve descartar imediatamente outras possíveis causas de sangramento e baixa contagem de plaquetas, como doenças crônicas ou efeitos colaterais de medicamentos que está tomando.
O especialista fará perguntas sobre seu histórico médico, realizará um exame médico inicial e solicitará um ou mais exames diagnósticos padrão. Estes últimos incluem:
As plaquetas são produzidas dentro da medula óssea, o tecido mole e esponjoso localizado no centro dos ossos grandes. Em alguns casos, o médico pode remover uma amostra do tecido ósseo e da medula óssea dentro dele por meio de uma biópsia. Alguns especialistas preferem colher amostras para análise por meio de um procedimento denominado aspiração, em que apenas parte do componente líquido é retirado da medula óssea. Na maioria das vezes, ambos os procedimentos são realizados simultaneamente e são chamados coletivamente de teste de medula óssea.

Na púrpura trombocitopênica idiopática, a medula óssea está livre de anormalidades porque a diminuição dos níveis de plaquetas é causada pela destruição de células na corrente sanguínea e disfunção do baço, e não por problemas na produção de plaquetas.
A púrpura trombocitopênica leve em adultos, cujas causas não foram estabelecidas de forma confiável, geralmente não requer tratamento específico - basta consultar um médico regularmente e verificar os níveis de plaquetas de vez em quando. As crianças quase nunca necessitam de terapia e o distúrbio desaparece por si só. Em adultos, contudo, pode ser necessário um tratamento eficaz se a condição for grave ou prolongada (crónica).
As opções de tratamento variam, desde medicamentos para aumentar a contagem de plaquetas até cirurgia para remover o baço (esplenectomia). É aconselhável discutir antecipadamente as opções de tratamento com seu médico, juntamente com os prós e contras da cirurgia. Alguns pacientes acreditam que os efeitos colaterais do uso de medicamentos lhes causam mais transtornos e desconforto do que as próprias consequências da doença.
Seu médico discutirá com você os efeitos colaterais e a dosagem dos medicamentos e suplementos dietéticos que você está tomando atualmente. É possível que alguns deles contribuam para a diminuição do nível de plaquetas no sangue circulante. Aspirina, ibuprofeno, Ginkgo biloba e varfarina têm esses efeitos colaterais.

Quando o diagnóstico de púrpura trombocitopênica é confirmado em adultos, o tratamento pode consistir nos seguintes medicamentos:
1. Medicamentos que suprimem o sistema imunológico. Muitos pacientes recebem corticosteróides orais, como a prednisona, no início da terapia. Ao reduzir a atividade do sistema imunológico, este medicamento ajuda a aumentar a contagem de plaquetas. Assim que o nível geral atingir um nível aceitável, você poderá parar gradualmente de tomar o medicamento sob a supervisão de um médico. O curso do tratamento leva de duas a seis semanas.
O problema é que a púrpura trombocitopênica em adultos, cujos sintomas desapareceram após o uso de Prednisona, pode retornar após o término da terapia. Os médicos geralmente prescrevem um novo ciclo de corticosteróides, mas este tratamento não pode ser continuado indefinidamente devido aos graves efeitos colaterais associados a este tipo de medicamento. A sobredosagem pode resultar em catarata, aumento dos níveis de açúcar no sangue, aumento do risco de infecção e osteoporose (enfraquecimento dos ossos).
2. Injeções para aumentar o número total de células sanguíneas. Se os corticosteróides não ajudarem, os médicos prescrevem uma injeção de imunoglobulina. Este medicamento também é utilizado nos casos em que o paciente apresenta sangramento intenso que requer intervenção cirúrgica imediata. O efeito desaparece dentro de algumas semanas. Os possíveis efeitos colaterais incluem dores de cabeça, vômitos e pressão arterial baixa.
3. Medicamentos que aceleram a produção de plaquetas. Medicamentos como Romiplostim e Eltrombopag (Promacta) ajudam a medula óssea a produzir mais plaquetas. Os efeitos colaterais podem incluir dores de cabeça, tonturas, náuseas e vômitos.
4. Outros imunossupressores. "Rituximab" ("Rituxan") ajuda a reduzir a intensidade da reação anormal do sistema imunológico, como resultado da destruição das plaquetas normais. Os possíveis efeitos colaterais incluem erupções cutâneas, pressão arterial baixa, febre e dor de garganta.
Se a púrpura trombocitopênica progredir (em adultos, seguir um estilo de vida saudável, evitar álcool e tomar medicamentos às vezes não ajuda a eliminar o problema), seu médico poderá recomendar a cirurgia. elimina rapidamente a patologia, pois o cirurgião elimina o principal destruidor de plaquetas. Deve-se levar em consideração, porém, que você correrá risco de complicações pós-operatórias. Além disso, as pessoas sem baço correm constantemente um risco aumentado de contrair doenças infecciosas.

Se, por motivos de saúde ou outros motivos, você não puder pagar a cirurgia, o especialista poderá prescrever novos medicamentos - por exemplo, Azatioprina (Imuran, Azasan). No entanto, medicamentos mais potentes têm efeitos colaterais mais graves. A azatioprina, em particular, causa febre, dores de cabeça, náuseas, vómitos e dores musculares.
Você foi diagnosticado com púrpura trombocitopênica? Em adultos, a ocorrência desta patologia exige mudanças no estilo de vida. Como a doença é acompanhada por um risco aumentado de sangramento, você deve evitar esportes de contato potencialmente perigosos (boxe, artes marciais, futebol) e tratar seu próprio corpo com o máximo de cuidado possível.
Normalmente, a púrpura trombocitopênica se desenvolve primeiro em crianças de 2 a 6 anos (até 10 anos), independentemente do sexo. Nos adultos, a doença não é tão comum e as mulheres têm maior probabilidade de sofrer com ela.
Uma característica desta doença é uma diminuição no número de plaquetas no soro sanguíneo abaixo do nível de 100 x10 9 /l no contexto de formação suficiente na medula óssea e a presença de anticorpos na superfície das plaquetas e no sangue que causam sua destruição.
Dependendo da duração e da ciclicidade do curso da doença, existem várias formas de púrpura trombocitopênica:
1.
Apimentado.
2.
Crônica.
3.
Recorrente.
A forma aguda é caracterizada por um aumento no nível de plaquetas sanguíneas superior a 150x10 9 /l dentro de 6 meses a partir da data de desenvolvimento da doença, na ausência de recidivas (casos repetidos da doença) posteriormente. Se a recuperação dos níveis plaquetários demorar mais de 6 meses, é feito o diagnóstico de púrpura trombocitopênica crônica. Quando seu número cai abaixo do normal novamente após a recuperação, ocorre púrpura trombocitopênica recorrente.
 Com esta doença, observa-se o aparecimento de uma erupção cutânea manchada e com hematomas na pele e hemorragias nas membranas mucosas. Os elementos da erupção cutânea podem ser de tamanhos variados, assemelham-se externamente a hematomas, são indolores quando pressionados, estão localizados de forma assimétrica e podem aparecer sem trauma, mais frequentemente à noite. A cor da erupção varia: do azulado ao amarelo.
Com esta doença, observa-se o aparecimento de uma erupção cutânea manchada e com hematomas na pele e hemorragias nas membranas mucosas. Os elementos da erupção cutânea podem ser de tamanhos variados, assemelham-se externamente a hematomas, são indolores quando pressionados, estão localizados de forma assimétrica e podem aparecer sem trauma, mais frequentemente à noite. A cor da erupção varia: do azulado ao amarelo. As hemorragias podem ocorrer não apenas nas membranas mucosas da cavidade oral e nas amígdalas, mas também no tímpano, no corpo vítreo, na esclera e no fundo do olho. Raramente é possível uma hemorragia cerebral, o que piora significativamente o estado do paciente. Isso é precedido pelo aparecimento de tonturas e dores de cabeça, além de sangramento em outros órgãos.
Quando o nível de plaquetas diminui para menos de 50x10 9 /l, aparecem sangramentos nasais e gengivais, que são mais perigosos quando um dente é removido. Nesse caso, o sangramento ocorre imediatamente e geralmente não recomeça após parar. Em adolescentes com púrpura trombocitopênica, o sangramento uterino durante a menstruação representa um certo perigo.
O complexo de exames inclui os seguintes procedimentos de diagnóstico:
 A púrpura trombocitopênica idiopática (PTI) se desenvolve em crianças entre 2 e 8 anos de idade. Meninos e meninas correm o mesmo risco de desenvolver esta patologia. A PTI começa agudamente em crianças após doenças infecciosas (mononucleose infecciosa, doenças infecciosas bacterianas, varicela), vacinação ou trauma. Deve-se notar que a incidência começa sazonalmente: mais frequentemente na primavera.
A púrpura trombocitopênica idiopática (PTI) se desenvolve em crianças entre 2 e 8 anos de idade. Meninos e meninas correm o mesmo risco de desenvolver esta patologia. A PTI começa agudamente em crianças após doenças infecciosas (mononucleose infecciosa, doenças infecciosas bacterianas, varicela), vacinação ou trauma. Deve-se notar que a incidência começa sazonalmente: mais frequentemente na primavera. Em crianças menores de 2 anos, é registrada a forma infantil de púrpura trombocitopênica. Nesse caso, a doença começa de forma aguda, sem presença de infecção prévia, e é extremamente difícil: o nível de plaquetas cai abaixo de 20x10 9 /l, o tratamento é ineficaz e o risco de cronicidade da doença é muito alto.
As manifestações clínicas da PTI dependem dos níveis plaquetários. O início da doença é caracterizado pelo aparecimento de erupções cutâneas com hematomas e hemorragias leves nas membranas mucosas. Quando o nível de plaquetas diminui para menos de 50 x10 9 /l, podem ocorrer vários sangramentos (nasal, gastrointestinal, uterino, renal). Mas na maioria das vezes, grandes “hematomas” em locais de contusões atraem a atenção; pode haver hematomas durante injeções intramusculares (injeções). Um baço aumentado é característico. Um exame de sangue geral registra trombocitopenia (diminuição de plaquetas), eosinofilia (aumento do número de eosinófilos), anemia (diminuição de hemoglobina).
O tratamento da púrpura trombocitopênica idiopática visa reduzir a produção de anticorpos antiplaquetários e prevenir sua ligação às plaquetas.
 1.
Glicocorticosteróides.
1.
Glicocorticosteróides.
Com o uso prolongado e individualmente, cada paciente pode apresentar efeitos colaterais ao tomar glicocorticóides: aumento dos níveis de glicose no sangue e diminuição dos níveis de potássio, úlceras estomacais, diminuição da imunidade, aumento da pressão arterial, retardo de crescimento.
2. Imunoglobulinas para administração intravenosa:
Com o uso de imunoglobulinas podem ocorrer dores de cabeça, reações alérgicas, aumento da temperatura corporal a níveis elevados e calafrios. Para reduzir a gravidade dos efeitos indesejáveis, são prescritos Paracetamol e Difenidramina por via oral e Dexametasona por via intravenosa.
3.
Interferon alfa.
Indicado para púrpura crônica em caso de tratamento ineficaz com glicocorticóides. 2x106 unidades de interferon-alfa são injetadas sob a pele ou no músculo durante um mês, 3 vezes por semana, em dias alternados.
Muitas vezes durante o tratamento com interferon aparecem
Púrpura trombocitopênica idiopática (PTI) - doença que se caracteriza por uma diminuição significativa do nível de plaquetas no sangue, o que geralmente leva à síndrome hemorrágica, ou seja, aumento do sangramento. O termo “doença de Werlhof” também é usado em homenagem ao médico que descreveu pela primeira vez a doença. Também foi proposto o uso do termo “trombocitopenia imune primária”.
Anticorpos contra suas próprias plaquetas são formados no corpo do paciente, a taxa de destruição das plaquetas sanguíneas aumenta várias vezes e ocorre uma deficiência dessas células. É a PTI a causa mais comum de síndrome hemorrágica em pacientes observados por hematologistas.
Existem formas agudas e crônicas de PTI. As formas agudas (que constituem a maioria dos casos da doença em crianças) duram menos de 6 meses. As formas crônicas diferem na frequência das recaídas: desde PTI com recaídas raras até um curso recidivante contínuo.
A frequência da PTI é, segundo diversas fontes, de 5 a 20 casos por 100.000 pessoas. A doença ocorre tanto em adultos quanto em crianças de qualquer idade, mas a frequência depende da idade: adultos jovens de 20 a 40 anos são os mais afetados, e crianças e idosos têm menor probabilidade de adoecer. As raparigas e as mulheres sofrem de PTI várias vezes mais frequentemente do que os rapazes e os homens. Em crianças pequenas, é mais frequentemente observada uma forma aguda da doença, em adolescentes e adultos - uma forma crônica.
As causas e o mecanismo de desenvolvimento da doença ainda não são totalmente compreendidos. Mas sabe-se que a doença pode ser desencadeada por uma infecção viral anterior: infecção viral respiratória aguda, rubéola, sarampo, citomegalovírus (CMV), vírus Epstein-Barr (EBV), etc. uma série de medicamentos, estresse, sobrecarga física, exposição excessiva ao sol, etc. A PTI não é uma doença hereditária.
A PTI é caracterizada por síndrome hemorrágica, ou seja, sinais de aumento de sangramento. Via de regra, a doença ocorre repentinamente. Observam-se hemorragias subcutâneas: pontuais (petéquias) e maiores (equimoses), áreas de hemorragia podem se fundir. Contusões facilmente, especialmente nos braços e pernas. Podem ocorrer hemorragias nas membranas mucosas e sangramento delas. Sangramento do nariz, gengivas e sangramento uterino em meninas são comuns. O sangramento intestinal e a presença de sangue na urina são menos comuns. Se os níveis de plaquetas estiverem muito baixos, são possíveis complicações potencialmente fatais, como hemorragias cerebrais. A perda significativa de sangue com o desenvolvimento de anemia grave também é perigosa.
Por outro lado, se a diminuição da contagem de plaquetas não for muito significativa, a doença pode não apresentar sintomas evidentes e ser detectada apenas pelo resultado de um exame de sangue, pelo aparecimento de hematomas isolados e, nas meninas, também por mais tempo. e menstruação mais intensa.
O primeiro sinal de PTI é um nível baixo de plaquetas sanguíneas - trombocitopenia. Outras contagens sanguíneas permanecem normais. São observadas alterações no coagulograma, como aumento do tempo de sangramento. Porém, para um diagnóstico preciso, é necessário excluir outras causas de baixos níveis plaquetários: leucemia, síndrome mielodisplásica, anemia aplástica, lúpus eritematoso sistêmico e outras doenças autoimunes, etc. (células a partir das quais as plaquetas são formadas). ; isso significa que a deficiência de plaquetas não se deve à produção insuficiente, mas à sua destruição no sangue. Os testes geralmente detectam anticorpos contra plaquetas em quantidades significativas. Um baço aumentado não é típico de PTI.
No diagnóstico, pode ser útil estudar a história médica, incluindo infecções virais anteriores, pois, conforme mencionado acima, podem provocar o desenvolvimento de PTI.
A terapia para pacientes com PTI é baseada em uma abordagem individual. A necessidade de tratamento é determinada pela necessidade de atingir e manter um nível seguro de plaquetas - afinal, com trombocitopenia profunda, são possíveis hemorragias com risco de vida. A terapia específica também é determinada por fatores como grau de sangramento (gravidade da síndrome hemorrágica), doenças concomitantes, etc.
Por exemplo, se o nível de plaquetas não estiver muito baixo, não houver sinais de hemorragias na pele ou estiverem diminuindo gradualmente e não houver sangramentos graves, então pode ser recomendada simplesmente uma monitorização cuidadosa do paciente.
Em casos mais graves, porém, o tratamento é necessário. Os hormônios glicocorticóides (prednisolona, metilprednisolona, dexametasona) são frequentemente usados como primeira linha de terapia. Mas seu uso a longo prazo está associado a efeitos colaterais indesejados. Também pode ser utilizada a administração intravenosa de imunoglobulinas (Pentaglobina, Octagam, etc.), que previnem o aumento da destruição das plaquetas sanguíneas.
Se a doença recorrer constantemente e não responder à terapia, os pacientes podem ser aconselhados a remover o baço (esplenectomia). Este procedimento é bem-sucedido na maioria dos casos, mas está associado a certos riscos e leva à perturbação da defesa imunológica do organismo. Por isso, muitas vezes tentam evitá-lo, principalmente em crianças.
O "Enplate" (romiplostim) é um medicamento que nos últimos 10-15 anos revolucionou o tratamento de muitos pacientes com formas crônicas e recorrentes de PTI. “Enplate” estimula o aumento da produção de plaquetas, é altamente eficaz, alcança resultados rapidamente e é bem tolerado. Outro medicamento eficaz para a forma crônica da doença é o Revolade (eltrombopag). Ambos os medicamentos permanecem eficazes mesmo com uso prolongado e podem ser usados em ambulatório (“Enplate” é administrado por via subcutânea, “Revolade” é usado em comprimidos), mas o alto custo ainda limita seu uso.
Às vezes são utilizados outros medicamentos, como imunossupressores (MabThera, Azatioprina, etc.). Medicamentos angioprotetores podem ser usados para melhorar a condição dos vasos sanguíneos. Existem outras opções de tratamento que podem ser individualizadas. Não são recomendadas transfusões de plaquetas de doadores, exceto em situações de sangramento com risco de vida, uma vez que tais transfusões potencializam a formação de anticorpos contra plaquetas.
Pacientes com PTI devem observar certas restrições, principalmente durante períodos de exacerbação. Eles precisam evitar lesões (o que limita severamente a sua capacidade de praticar esportes); tomar aspirina e outros medicamentos que reduzem a coagulação sanguínea; contato com alérgenos, exposição excessiva ao sol, vacinações, etc.
As formas agudas de PTI geralmente desaparecem dentro de algumas semanas ou meses, pois os anticorpos plaquetários podem circular no sangue por até 6 meses. Na forma crônica da PTI, na maioria dos casos é possível alcançar uma remissão estável, mas às vezes, infelizmente, a doença segue um curso frequentemente recidivante. Porém, acredita-se que em média esta doença tenha um bom prognóstico, principalmente com o uso de medicamentos modernos.
A vasculite hemorrágica é uma doença acompanhada de processos inflamatórios de microvasos e ocorre com formação de trombos. Refere-se a vasculite sistêmica. Afeta órgãos internos e vasos sanguíneos - arteríolas, vênulas e capilares. Na maioria das vezes, os intestinos e os rins sofrem de patologia.
De acordo com a CID-10, trata-se de púrpura alérgica. Desenvolve-se principalmente em crianças de 5 a 14 anos, ocorrendo em 23 a 25 pessoas em cada 10 mil. Foram registrados casos isolados da doença que afetam crianças menores de 3 anos.
Quando afetados por vasculite, aparecem anticorpos imunológicos no sangue, que durante a circulação são depositados nas paredes dos vasos sanguíneos e causam danos locais. Processos inflamatórios assépticos se formam nas áreas afetadas, o que aumenta a permeabilidade vascular. A fibrina é depositada no lúmen dos vasos, desenvolve-se microtrombose e surge a síndrome hemorrágica.
A vasculite hemorrágica em adultos, ao contrário do curso da doença em crianças, é caracterizada por sintomas leves. A forma abdominal ocorre em metade dos casos.
A púrpura de Henoch-Schönlein é dividida em tipos:

A púrpura hemorrágica é classificada de acordo com a duração da doença:
A vasculite hemorrágica é dividida em 3 graus de atividade:

A doença costuma ser benigna. A partir do momento da primeira erupção cutânea, na maioria dos casos, a recuperação espontânea ocorre dentro de 2 a 3 semanas. Em alguns casos, é possível uma recaída - a doença reaparece depois de um tempo. Complicações graves ocorrem com danos aos rins e intestinos.
Existem vários cursos clínicos de vasculite hemorrágica.
A forma cutânea é expressa em todos os pacientes. Na pele aparecem erupções cutâneas difusas do tipo maculopapular, que não desaparecem com a pressão. O tamanho das erupções cutâneas costuma ser pequeno e localizado simetricamente; em alguns casos, aparece uma erupção cutânea urticariforme (tipo alérgico).
A abundância de manifestações cutâneas depende da gravidade do curso - na pior das hipóteses, necrose e úlceras podem se desenvolver nos locais de formação da erupção cutânea. As erupções cutâneas aparecem na área das grandes articulações, coxas, nádegas e pernas. Raramente - nos braços e no tronco. Na vasculite crônica, especialmente se as recidivas forem comuns, a pele no local da erupção cutânea descama. Hiperpigmentação é observada.
A síndrome articular aparece em 70% dos pacientes. Os danos ao tecido articular podem durar vários dias ou ser de curto prazo. Aparece dor nas articulações, vermelhidão e inchaço são pronunciados. Nesta forma da doença de Henoch-Schönlein, os sintomas limitam a mobilidade da articulação, dificultando muito os movimentos. As articulações maiores são afetadas com mais frequência, especialmente os tornozelos ou joelhos. Esta síndrome ocorre no estágio inicial ou final da doença. Não leva a problemas articulares crônicos.

A forma abdominal precede ou ocorre junto com a forma articular da vasculite hemorrágica; os sintomas se manifestam por dor abdominal moderada ou paroxística. Muitas vezes o paciente não consegue indicar a localização exata da dor. Evacuações intestinais anormais, vômitos e náuseas são possíveis. Os sinais de dor geralmente desaparecem espontaneamente ou alguns dias após o tratamento. Nos casos graves da doença, pode ocorrer sangramento gastrointestinal com presença de sangue no vômito e nas fezes.
A síndrome renal é caracterizada como um curso de glomerulonefrite aguda ou crônica. Ocorre em 20-30% dos pacientes. A síndrome nefrótica pode se desenvolver. Pode causar insuficiência renal.
Outros órgãos raramente são afetados pela doença. Possível pneumonia hemorrágica, que ocorre com danos aos pulmões. Miocardite hemorrágica ou pericardite com danos ao tecido cardíaco. Meningite hemorrágica com sintomas de lesão cerebral - aparecem dor de cabeça, sensibilidade à luz, irritabilidade, tontura e pode causar acidente vascular cerebral hemorrágico (sangramento no cérebro).
O diagnóstico da púrpura de Henoch-Schönlein é realizado por um reumatologista. A faixa etária do paciente, os dados clínicos são levados em consideração e os exames laboratoriais são realizados. A anamnese é coletada para determinar a presença de outras doenças. Nos exames de sangue em pacientes com vasculite hemorrágica, o processo inflamatório e o aumento do número de plaquetas e eosinófilos são mais frequentemente expressos.

O exame de urina mostra aumento do conteúdo proteico, hematúria e cilindrúria. Um aumento na imunoglobulina A e PCR é observado nos parâmetros bioquímicos do sangue. Um coagulograma é realizado para determinar as leituras de coagulação sanguínea.
Se a função renal estiver prejudicada, você deve consultar um nefrologista. Eles fazem uma ultrassonografia dos rins e uma ultrassonografia de seus vasos. Um teste de urina é feito para estudos bioquímicos e um teste de Zimnitsky.
No caso da forma abdominal, excluem-se outras doenças que podem causar os mesmos sinais sintomáticos de “abdome agudo”. Estes incluem apendicite, pancreatite, penetração de úlceras gastrointestinais, colecistite, etc. Um cirurgião gastroenterologista escreve um encaminhamento para ultrassonografia abdominal e gastroscopia. Um exame de sangue oculto nas fezes é feito para diagnosticar um possível sangramento gastrointestinal.
Em casos graves da doença, é realizada uma biópsia de pele. Estudos histológicos revelam depósitos de imunoglobulina A e CIC nas paredes dos capilares, vênulas, arteríolas e microtrombose com vazamento de sangue para fora dos vasos. O diagnóstico diferencial da vasculite hemorrágica é feito com outras doenças que podem causar sintomas semelhantes.
Na fase aguda, no tratamento da vasculite hemorrágica, são prescritos repouso no leito e dieta com restrição de alimentos alergênicos. Você não deve tomar antibióticos ou outros medicamentos que aumentem a sensibilidade a agentes estranhos no sangue.
A heparina é o principal medicamento utilizado no tratamento da doença de Henoch-Schönlein. São prescritos corticosteróides, se a medida for ineficaz, são utilizados citostáticos. A prednisolona também é usada em alguns casos, mas as opiniões sobre a eficácia do medicamento no tratamento da púrpura de Henoch-Schönlein estão divididas. Nos casos graves da doença, são prescritas imunossorção, hemossorção e plasmaférese.

O tratamento da vasculite hemorrágica na síndrome articular é feito com antiinflamatórios (ibuprofeno ou indometacina). Os anti-histamínicos são considerados ineficazes e são usados para aliviar as manifestações alérgicas. Os enterosorbentes são prescritos em casos de doenças abdominais e alergias alimentares.
A terapia de infusão é realizada diluindo o sangue circulante e reduzindo a concentração de possíveis alérgenos e anticorpos imunológicos. Use com cautela em pacientes com doença renal. Os medicamentos necessários para o procedimento de tratamento da doença são selecionados individualmente. São utilizadas soluções cristalóides - eletrólito concentrado com açúcares diluídos.
As crianças devem estar registradas em um dispensário por 2 anos. Durante os primeiros 6 meses, o paciente visita o médico assistente todos os meses. Depois disso - uma vez a cada 3 meses, então - uma vez a cada 6 meses. Mais frequentemente, o prognóstico para a eficácia do tratamento da vasculite hemorrágica é favorável.
A prevenção é feita higienizando os focos de infecção. Amostras de fezes são coletadas todos os dias para verificar a presença de ovos de helmintos. As crianças não devem fazer fisioterapia; o esforço físico excessivo e a exposição prolongada ao sol são contra-indicados. A vasculite hemorrágica em adultos é mais leve do que em crianças.

Suco de laranja. O significado simbólico do suco de laranja nos livros de sonhos é prazer e tentação. Muitas vezes nós...

Na maioria das vezes superamos todos os tipos de dificuldades encontradas ao longo do caminho da vida. Claro, para isso fazemos...

Este ano o seu patrono Netuno estará na sua constelação e isso é um bom sinal, pois você se sentirá...

1993 quem? 1993 é o ano de qual animal? — De acordo com o horóscopo chinês, 1873, 1933, 1993 pertenciam aos anos da Água Negra...

A dualidade onda-partícula da luz significa que a luz tem simultaneamente as propriedades de eletromagnética contínua...

O papel da biologia é enorme em nosso mundo. Embora não seja um assunto prioritário, a maioria dos alunos e pais...
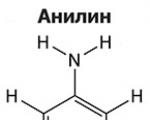
As aminas são derivados orgânicos da amônia contendo um grupo amino NH 2 e um radical orgânico. Em geral...
Como responder às perguntas da Parte BA segunda parte do trabalho de estudos sociais consiste em 7 tarefas com uma resposta curta....

O formulário TORG-15 é elaborado no caso de durante o transporte, movimentação entre e dentro do armazém, quando...

Os nutricionistas dizem que para ter uma boa saúde e um corpo esguio é preciso incluir lanches na sua...

Deliciosas cenouras em conserva para o inverno podem ser preparadas em vários recipientes, pode ser de madeira...

Quando me restam alguns minutos para preparar o café da manhã, usam-se as receitas mais simples e rápidas....

Uma mesa elegante, uma árvore de Natal decorada, espírito de tangerina espalhado por todos os quartos, em breve o feriado mais mágico -...

Cada um de nós enfrentou repetidamente dificuldades financeiras e períodos difíceis na vida quando a Fortuna...

Na maioria das vezes superamos todos os tipos de dificuldades encontradas ao longo do caminho da vida. Claro, para isso nós...

Este ano o seu patrono Netuno estará na sua constelação e isso é um bom sinal, pois você...