Що можна і не можна на Різдвяний піст?
У 2018 році Різдвяний пост розпочнеться 28 листопада. У цей період православні віруючі готуються до зустрічі Різдва.

Тирозинкіназа Брутона
Хвороба Брутона - Спадкова патологія, що характеризується дефіцитом гуморального імунітету. Вроджена хвороба пов'язана з мутацією гена, розташованого на Х-хромосомі. Дефект гена супроводжується порушенням формування та функціонування В-лімфоцитів, нейтрофілів, тромбоцитів та інших клітин крові, що зароджуються у кістковому мозку.
Синдром Брутона є першим у світі вивченим спадковим імунодефіцитом. Вперше був виявлений 1952 року педіатром Огденом Карр Брутоном, який вивчав рецидивні інфекції пневмонії у 8-річного хлопчика. Лікар з'ясував, що дитина перенесла ще ряд бактеріальних захворювань і встановила причину — дефіцит гамма глобулінів у крові, тобто наявність агаммаглобулінемії.
Брутон був першим лікарем, який забезпечив специфічну імунотерапію своєму маленькому пацієнту за допомогою ін'єкцій IgG імуноглобуліну. Генетичну природу агаммаглобулінемії вченим вдалося з'ясувати в 1993 році, дефект гена отримав назву Тирозінкіназа Брутона.

Наслідування при хворобі Брутона
Мутація в гені передається дітям обох статей, але проявляється лише у хлопчиків, а дівчатка можуть стати носіями. Дитина чоловічої статі успадковує дефект, якщо його мати є носієм, а батько здоровий, дівчатка в цьому випадку можуть отримати дефектний ген у 50% випадків. Коли у матері відсутні мутації в гені, а батько хворий – сини народжуються здоровими, дівчатка успадковують захворювання. Хвороба Брутона зустрічається із частотою 1:250 000 дітей чоловічої статі.
Причина агаммаглобулінемії полягає в численних мутаціях (понад 1000) гена цитоплазматичної тирозинкінази. Найбільший вплив тирозинкіназа на дозрівання В-лімфоцитів - захисних клітин організму, хоча присутній і в інших клітинах крові, але механізм впливу мутації на них до кінця не з'ясований. Передбачається, що інші ферменти замінюють тирозинкіназу в нейтрофілах, тромбоцитах, моноцитах та інших клітинах. У Т-лімфоцитах Тирозинкіназа Брутона не виявлено, тому зазвичай Т-лімфоцити розвиваються та функціонують у природному режимі або підвищують свою активність. На відміну від інших патологій гуморального імунітету, при захворюванні Брутона відзначається нестача імуноглобулінів усіх класів.

«Поломка» при хворобі Брутона
Більшість порушень гена тирозинкінази - це точкові мутації, що провокують аномалії білка, необхідного для формування та диференціювання В-лімфоцитів. В-лімфоцити здатні зароджуватись, але не досягають зрілих форм, тобто не можуть виробляти антитіла. Дефіцит цих клітин призводить до нездатності організму забезпечити оптимальну імунну відповідь проти бактерій та інших патогенних мікроорганізмів. Патологічні відхилення в процесі дозрівання імунних клітин можуть проявлятися як значним зниженням, так і повною відсутністю В-лімфоцитів та антитіл у крові.
Особливу сприйнятливість пацієнти відчувають до специфічних бактерій - стафілококів, пневмококів, менінгококів, гемофільних прокаріотів. До вірусних інфекцій спостерігається більш висока стійкість у ранньому віці, проте згодом організм не в змозі протистояти і вірусам. Ближче до підліткового віку нерідко проявляється системне захворювання, спричинене ентеровірусною інфекцією. Загалом пацієнти схильні до аутоімунних, онкологічних, суглобових патологій, страждають від алергічних реакцій.

Початок проявів захворювання – дитячий вік
Ознаки захворювання можуть виникнути у віці кількох місяців, коли організм дитини перестає отримувати імунні антитіла від матері, іноді хвороба виявляється у віці 2-3 років. Розвиваються рецидивні інфекційні зараження, які різною мірою і варіабельності зберігаються протягом усього життя. Хвороби протікають важче і довше, ніж зазвичай, переходячи у хронічні стадії. Характерна наявність гнійних інфекцій, які вражають різні органи.
Відзначаються часті гнійно-запальні ураження приносових пазух, вух, слизової оболонки очей. Бронхи незворотно змінюються внаслідок хронічного нагноєння, що проявляється задишкою, хрипким кашлем, відчуттям нестачі повітря. Суглоби часто опухають, викликаючи періодичні напади болю. У третини пацієнтів утворюються артрити великих суглобів.
Мигдалики дуже малі, лімфатичні вузли невеликого розміру, при інфікуванні організму вони не збільшуються. Шкірні покриви уражаються поверхневими та глибокими формами стрептодермії. Постійні інфекційні ураження нерідко супроводжуються хронічною діареєю, запальними явищами у кишечнику та тканинах інших органів.
Синдром Брутона іноді супроводжується порушенням слуху, зору. Найчастішою ознакою є пародонтит. Дитина може відставати у рості, і навіть мати дефіцит маси тіла. Інтелект захворювання не впливає, у деяких випадках у пацієнтів з раннього віку відзначені видатні розумові здібності.

Картина крові має свої характеристики
При діагностиці вивчається анамнез пацієнта, за допомогою рентгенографії відзначаються характерні ознаки: аномально малі розміри мигдаликів та лімфовузлів, порушення будови селезінки.
Дані лабораторного дослідження виявляють:
Додаткові обстеження включають УЗД органів черевної порожнини, колоноскопію, діагностику легень.

Пацієнтам показано замісну терапію.
Пацієнтам необхідна постійна замісна терапія протягом усього життя. Для цього використовуються ін'єкції імуноглобулінів, починаючи з введення IgG у високій концентрації до дози насичення, потім дозування знижується. Частота та доза введення імуноглобулінів визначається індивідуально. Зазвичай пацієнт повинен одержувати імуноглобуліни раз на 3 тижні у кількості від 300 до 500 мг/кг ваги. Ще одним методом терапії є переливання плазми від здорових донорів.
На тлі інфекційних захворювань дозу імуноглобулінів підвищують, обов'язковим елементом лікування є масивне введення антибіотиків. Антибіотики призначаються залежно від інфекції, але тривалість їх прийому пролонгована, а доза максимальна. У деяких випадках антибактеріальна терапія проводиться щодня, навіть за відсутності інфікування.
Профілактика полягає у відвідуванні генетика перед зачаттям дитини. Якщо дитина народилася із хворобою Брутона, необхідно починати терапію з перших днів життя. Вакцинацію на основі живих вірусів (поліомієліт, кір, епідемічний паротит, краснуха) виключено. Хлопчики у тій самій сім'ї, зокрема двоюрідні, повинні бути обов'язково обстежені на наявність патології.
Для підтримки нормальної життєдіяльності крім терапії слід дотримуватись правил: уникати контакту з хворими людьми, проводити якнайбільше часу на чистому повітрі, повноцінно харчуватися та відпочивати.

Рання діагностика та своєчасне лікування – сприятливий прогноз
Прогноз залежить від того, наскільки рано було виявлено патологію та від супутніх захворювань. Зв'язку між тією чи іншою мутацією в гені тирозинкінази та тяжкістю захворювань не виявлено. Нерідко терапія імуноглобулінами та активне лікування антибіотиками не можуть запобігти розвитку важких форм пневмоній, менінгітів, ракових пухлин або лейкемії.
Однак у періоди відсутності інфекційних захворювань пацієнти здатні вести активний спосіб життя, займатися спортом, вчитися чи працювати. При коректному веденні захворювання випадки інфікування зводяться до 3-4 разів на рік.
Надією для пацієнтів є розробки в галузі генної терапії для корекції мутації тирозинкінази Брутона.
РОЗДІЛ 3. ІМУНОДЕФІЦИТНІ СТАНИ
Визначення, клінічні прояви та класифікація імунодефіцитних станів. Класифікація первинних ІДС. Первинні ІРС адаптивного імунітету з переважним ураженням В-клітинної ланки. Первинні ІРС адаптивного імунітету з переважним ураженням Т-клітинної ланки. Комбіновані Т-і В-імунодефіцити. Первинні дефіцити фагоцитарної ланки уродженого імунітету. Первинні дефіцити системи комплементу. Попередня діагностика первинних ІДС. Визначення, основні причини формування вторинних імунодефіцитів. Фізіологічні (вікові) імунодефіцити. Роль попередніх захворювань, ятрогенних факторів, несприятливих екологічних факторів та факторів ризику способу життя у виникненні вторинних ІДС.
У функціонуванні імунної системи, як і будь-якої іншої системи організму, можуть виникати порушення, що ведуть до розвитку захворювань, характерних насамперед для цієї системи. Одним із найпоширеніших варіантів таких порушень є імунодефіцитні стани (ІДС), що характеризуються нездатністю імунної системи розвивати нормальну імунну відповідь.
Імунодефіцитні стани (ІДС) – це зниження функціональної активності основних компонентів імунної системи, що веде до порушення імунологічного реагування, до зниження або формування неадекватної вродженої або адаптивної імунної відповіді на різні патогени та антигени, в першу чергу – на інфекційні.
Необхідно відрізняти поняття імунодефіцит від поняття імунологічної толерантності, за якої також знижено або відсутня імунна відповідь. На відміну від імунодефіциту, імунологічна толерантність завжди специфічна, тобто формується тільки проти одного конкретного антигену або групи антигенів. При імунодефіцитних станах, зазвичай, знижується імунологічна реактивність загалом.
Наявність ІДС насамперед призводить до зниження протиінфекційного захисту організму, що клінічно проявляється у підвищенні інфекційної захворюваності.
Для ІДС з переважним ураженням В-клітинної ланки адаптивного імунітетухарактерно виникнення рецидивуючих інфекцій, у тому числі викликаних стафілококами, стрептококами, пневмококами, збудниками кишкових інфекцій та ін.
При ІДС з переважним ураженням Т-клітинної ланки адаптивного імунітетуна додаток до багатьох бактеріальних інфекцій виникають інфекційні захворювання, викликані такою умовно-патогенною флорою, як гриби Candida, віруси (герпес, цитомегаловірус, аденовіруси), пневмоцисти, внутрішньоклітинні бактерії (мікобактерії, уреаплазми та ін.). Характерна також схильність до генералізації туберкульозної інфекції, підвищено частоту гельмінтозів.
Первинні ІДС можуть бути зумовлені дефектами компонентів вродженого імунітету. Ці дефекти можуть зачіпати різні ланки системи вродженого імунітету: компоненти та інгібітори системи комплементу, дефекти фагоцитарних клітин, NK-клітин, рецепторів і транспортних молекул (ТАР), що беруть участь у процесингу антигенів. У зв'язку з інтенсивними дослідженнями в цій галузі, які проводяться із застосуванням сучасних генетичних та молекулярно-біологічних методів, виявляються нові варіанти первинних ІДС вродженого імунітету, а також поєднаних ІДС вродженого та адаптивного імунітету. Як правило, первинні ІДС вродженого імунітету виявляються у вигляді різних інфекційних захворювань, наприклад:
· Дефіцит компонентів комплементу(С1, С4, С2; С3, С5-9 та ін) призводить до рецидивуючих генералізованих інфекцій, викликаних інкапсульованими мікроорганізмами (Neisseria meningitidis, пневмокок, гемофільна паличка). Часто зустрічається системний червоний вовчак.
· Дефекти фагоцитарної системиклінічно частіше виявляються у вигляді інфекційних уражень шкіри та паренхіматозних органів, наприклад, викликаних стафілококами та клебсієлами при хронічній гранулематозній хворобі.
До клінічних проявів ІДС належать також гематологічні порушення (лімфоаденопатії, лейкози), характерне виникнення уражень шлунково-кишкового тракту, зумовлених значною мірою порушенням місцевого імунітету ШКТ. Часто ІДС супроводжуються виникненням аутоімунних та алергічних захворювань. Особливе місце належить онкологічним захворюванням. Найчастіше у хворих на ІДС зустрічаються гемобластози, лімфопроліферативні захворювання, саркома Капоші.
Усі імунодефіцити прийнято розділяти на первинні, чи вроджені, і вторинні, чи придбані.Вторинні ІДС не є генетично детермінованими, такі дефекти імунної системи набуваються протягом життя і є наслідком будь-яких впливів на імунну систему або наявності інших захворювань.
Первинні імунодефіцитні стани
Первинні ІДС – це спадкові порушення вродженої та/або адаптивної імунної системи, пов'язані з генетичними дефектами одного або кількох компонентів. Первинні ІДС адаптивної імунної системиобумовлені переважними дефектами її В-клітинної та Т-клітинної ланки, у багатьох випадках мають місце поєднані ураження обох ланок. Найбільш вивченими первинними ІДС уродженого імунітетує ІДС, зумовлені генетичними дефектами системи комплементу та фагоцитарної системи.
При нормальному перебігу вагітності у внутрішньоутробному періоді розвитку дитина перебуває у стерильних умовах. Негайно після народження організм починає заселятися мікроорганізмами. Оскільки мікрофлора, що оточує людину, не є патогенною, ця колонізація не викликає хвороби. Надалі зустріч із патогенними мікроорганізмами, із якими дитина ще зустрічався, викликає розвиток відповідної інфекційної хвороби. Кожен контакт із патогеном призводить до розширення імунологічної пам'яті та формує довготривалий імунітет.
У захисті організму від постійних атак патогенних вірусів, бактерій, грибів та найпростіших беруть участь два основних компоненти вродженого імунного захисту: фагоцитоз і комплемент, а також два компоненти адаптивного захисту: В-клітинний (антитіла) та Т-клітинний. Кожен із цих компонентів може функціонувати незалежно, але найчастіше вони працюють у тісному комплексному взаємодії. Вроджені дефекти однієї із зазначених ланок призводять до порушення захисту організму загалом і клінічно проявляються у вигляді різних варіантів первинних імунодефіцитів.
В даний час завдяки ідентифікації молекулярних порушень, що лежать в основі патогенезу багатьох первинних імунодфіцитів, і у зв'язку з великою варіабельністю клінічної картини стало ясно, що цей вид патології набагато більш поширений, ніж вважалося раніше.
Рання діагностика та адекватне лікування є умовою життя пацієнтів із багатьма формами первинних імунодефіцитів. Без своєчасної правильної діагностики та адекватного лікування дитина гине, як правило, протягом першого року життя. Кожен вид імунодефіциту має свою вже відпрацьовану тактику лікування, і більшість первинних дефектів імунної системи в даний час підлягає корекції. Своєчасна діагностика та лікування первинних імунодефіцитів є одним із невикористаних резервів для зниження захворюваності, смертності та інвалідності населення загалом.
Первинні ІДС, як правило, мають спадкову природу,тому їх ще називають генетично обумовленими ІРС. Найчастіше первинні ІДС успадковуються за рецесивного типу. У тих випадках, коли генетичні дефекти зачіпають специфічні фактори імунітету (антитілоутворення, клітинні форми адаптивної імунної відповіді), їх називають ще специфічними ІДС, на відміну від дефектів неспецифічних компонентів захисту (фагоцитозу, системи комплементу та ін.).
Іноді стосовно первинних ІРС застосовують термін «вроджені», що передбачає можливість виникнення захворювання на основі спадкових дефектів, що виникли в ембріональному періоді, наприклад, селективний дефіцит IgA як наслідок внутрішньоутробно перенесеної корової краснухи.
Первинні імунодефіцити різноманітні, та його опис і класифікація у літературі постійно змінюються. Однією з перших класифікацій імунодефіцитів адаптивної імунної системи є запропонована 1974 року Ю.М. Лопухіним та Р.В. Петровим класифікація, основою якої покладено рівні генетичних блоків різних етапах диференціювання Т- і В-лімфоцитів.
Відповідно до цієї класифікації, все різноманіття форм ІДС адаптивної імунної системи поділено на три групи:
1. Первинні ІРС з переважним ураженням Т-системи (блок II, VI).
2. Первинні ІРС з переважним ураженням B-системи (блок III, IV, V).
3. Комбіновані первинні ІРС з одночасним ураженням Т-і В-систем (блок I, блоки V+VI та ін.).
 I
I
Стовбурова III
кровотворна
|
|
|
Малюнок 42. Патогенетична схема класифікації первинних ІДС Т- та В-систем імунітету
(Р.В. Петров, Ю.М. Лопухін)
Блок I. Практично відсутні стволові кровотворні клітини. Характерна генералізована аплазія кровотворної та лімфоїдної тканини. Чи не утворюються ні Т-, ні В-лімфоцити.
До цього блоку відносили важку комбіновану імунологічну недостатність (ТКІН), яка зустрічається у різних формах.
Блок ІІ. Блок на ранніх етапах розвитку внутрішньотімусних Т-лімфоцитів. Повне вимкнення адаптивного клітинного імунітету. У таких хворих характерно виникнення тяжких рецидивуючих вірусних інфекцій, що призводять до загибелі у ранньому (до 1 року) віці; висока частота вроджених аномалій розвитку; у 100-1000 разів вищий за ризик виникнення злоякісних пухлин; організм пацієнта не відкидає чужорідний трансплантат.
Цей блок представлений, зокрема, аплазією чи гіпоплазією тимусу (синдром Ді Джорджі).
Блок ІІІ. Для цього блоку характерна агаммаглобулінемія, зчеплена з Х-хромосомою (хворіють на хлопчики). У таких дітей антитіла практично не виробляються, що призводить в першу чергу до частих бактеріальних інфекцій, що важко протікають. Цей варіант імунодефіциту називається хворобою Брутона, він діагностується, як правило, у другому півріччі життя, оскільки протягом перших місяців організм дитини захищений материнськими антитілами, що пройшли через плаценту і містяться в материнському молоці.
Блок IV. Дещо знижено число В-лімфоцитів. Гіпогаммаглобулінемія з макроглобулінемією, оскільки синтез IgM збережений або навіть підвищений, тоді як рівень IgG та IgA різко знижений.
Блок V Селективний дефіцит IgA. Через відсутність основного секреторного імуноглобуліну у таких дітей розвиваються насамперед інфекційні захворювання, пов'язані зі слизовими оболонками носоглотки, дихальних шляхів, ШКТ, урогенітального тракту.
Блок VI. Зачіпає процеси дозрівання та виходу Т-лімфоцитів у периферичні лімфоїдні органи та кров'яне русло. Характерна поєднана патологія з переважним ураженням функцій Т-клітинного імунітету.
Нині ідентифіковано понад 70 вроджених дефектів як уродженої, і адаптивної імунної системи. Ймовірно, у міру вдосконалення методів молекулярної імунодіагностики їх кількість зростатиме. Ендогенні, як правило, генетично обумовлені дефекти одного з компонентів імунної системи призводять до порушення всієї системи захисту організму та клінічно виявляються як одна з форм первинного імунодефіцитного стану. Так як при нормальному функціонуванні імунної системи в комплексній імунній відповіді беруть участь багато типів клітин та сотні молекул, в основі патогенезу клінічних форм первинних ІДС лежать численні варіанти дефектів.
В останні роки прийшло усвідомлення того факту, що клінічні прояви первинних ІДС можуть спостерігатися не тільки у новонароджених, а й у пізнішому віці. Це пов'язано з тим, що дефект однієї з ланок імунітету може якийсь час не виявлятися клінічно у вигляді підвищеної інфекційної захворюваності, тому що всі інші компоненти імунітету збережені і компенсують цей дефект, поки не виснажені їх резервні можливості.
Первинні ІДС - це рідкісні захворювання, їх частота в середньому становить 1/25000 - 1/100000, хоча селективний дефіцит IgA зустрічається набагато частіше: 1/500 - 1/700 осіб. За даними Європейського регістру первинних ІРС за 1997 рік частота зареєстрованих первинних ІРС в середньому по Європі становила 1/96000 населення, тоді як в окремих країнах вона була суттєво вищою: 1/38000 (Великобританія); 1/12500 (Швейцарія); 1/10 000 (Швеція). Швидше за все такі відмінності обумовлені рівнем розвитку медицини в різних країнах, і реальна частота первинних ІДС істотно вище за середні цифри, що публікуються.
В даний час загальноприйнятою є наступна спрощена класифікація первинних ІДС:
I. Первинні ІДС з переважним ураженням гуморального адаптивного імунітету
1. Зчеплена з Х-хромосомою агама (гіпогама) глобулінемія – хвороба Брутона.
2. Загальна варіабельна імунологічна недостатність (ОВІН) – загальна варіабельна гіпогаммаглобулінемія.
3. Транзиторна гіпогамаглобулінемія у дітей (повільний імунологічний старт).
4. Виборчий дефіцит імуноглобулінів (селективний дефіцит IgA).
ІІ. Первинні ІДС з переважним ураженням Т-клітинного адаптивного імунітету
1. Синдром Ді Джорджі (гіпо-, аплазія тимусу).
2. Хронічний слизово-шкірний кандидоз.
ІІІ. Комбіновані Т- та В-імунодефіцити адаптивного імунітету
1. Тяжка комбінована імунологічна недостатність (ТКІН):
а) Х-зчеплена;
б) аутосомно-рецесивні.
2. Атаксія - телеангіектазія (синдром Луї-Барр).
3. Синдром Віскотта-Олдріча.
4. Імунодефіцит із підвищеним рівнем IgM (зчеплений з Х-хромосомою).
5. Імунодефіцит з карликовістю.
IV. Дефіцити фагоцитарної системи
1. Хронічний лімфогранулематоз.
2. Синдром Чедіака-Хігассі.
3. Синдром гіпер-IgE (синдром Джоба).
V. Дефіцити системи комплементу
1. Первинні ІРС, обумовлені дефіцитом компонентів комплементу.
2. Первинні ІРС, обумовлені дефіцитом інактиваторів системи комплементу.
Первинні ІДС з переважним ураженням
гуморального адаптивного імунітету
Хвороба Брутона
Вперше в 1952 році американський педіатр Брутон (Bruton) описав 8-річного хлопчика, який страждав на різні інфекційні захворювання, який до 4-річного віку 14 разів хворів на пневмонію, неодноразово отити, синусити, переніс сепсис і менінгіт. При дослідженні у нього у сироватці крові не виявили антитіл. Це був перший опис первинного імунодефіциту у науковій медичній літературі, виділеного як самостійна нозологічна форма.
Хвороба Брутона має рецесивний тип успадкування, зчеплений із Х-хромосомою, хворіють на неї лише хлопчики. Захворювання також позначається як Х-зчеплена агаммаглобулінемія. Зустрічається із частотою 1/1000000 населення. У Великій Британії частота цього імунодефіциту становить 1/100000.
В основі хвороби Брутона лежить дефект ферменту цитоплазматичної тирозинкінази, який здійснює передачу сигналу в ядро В-лімфоцитів для його активації з подальшим перетворенням на плазматичну клітину. Тим самим стає неможливим синтез імуноглобулінів.
Перші ознаки захворювання проявляються починаючи з 7-8 місяців до 2-3 років. Трансплацентарне перенесення імуноглобулінів зазвичай забезпечує хворих цією хворобою дітей кількістю антитіл, достатньою для того, щоб уникати інфекцій протягом декількох перших місяців життя. Надалі відсутність синтезу антитіл призводить, як правило, до виникнення рецидивуючих бактеріальних інфекцій, переважно дихальних шляхів та шкіри. Зазвичай збудниками є стрептококи, стафілококи та грамнегативні бактерії. Інфекції мають тенденцію поширюватися, що призводить до септицемії та менінгіту. Однак у таких дітей зберігається стійкість до вірусних інфекцій, оскільки клітинний імунітет не порушено.
При об'єктивному оглядізнаходять незвичайно маленькі, гладкі мигдалики, лімфатичні вузли невеликі, селезінка не збільшена. Звертає особливу увагу той факт, що лімфовузли, печінка та селезінка не реагують збільшенням на інфекційно-запальний процес в організмі, що є важливою діагностичною ознакою.
У слизовій оболонці кишечника повністю зникають плазматичні клітини, хоча в нормі вони там утримуються в досить великій кількості. У зв'язку з цим спостерігаються порушення всмоктування, часто розвиваються хронічні ентерити. У кишечнику часто виявляють абсцеси зі скупченням лейкоцитів, що розпадаються, в криптах кишки.
При лабораторному обстеженніу периферичній крові відзначається різке зниження або відсутність В-лімфоцитів, низькі рівні або відсутність усіх класів імуноглобулінів.
Лікування:постійна замісна терапія імуноглобулінами. Введення препаратів імуноглобулінів проводиться внутрішньовенно по 200-600 мг/кг на місяць. У разі виникнення інфекційного захворювання додатково вводять специфічні імуноглобуліни (антистафілококовий, антистрептококовий, протикоровий і т.д.).
Прогноз:при правильно поставленому діагнозі та своєчасно розпочатому лікуванні відносно сприятливий.
Хвороба Брутонаабо Агаммаглобулінемія є варіантом імунодефіциту – гуморального первинного його різновиду. Викликається генною мутацією, яка кодує тирозинкіназу Брутона.
Хвороба Брутона є чисто хлопчиковим захворюванням, яке, через особливості хромосом в ДНК, дівчаток не вражає. Поширеність її – один випадок на чверть мільйона підлітків. Носіями гена, що є причиною проблеми, можуть бути жінки. Вони й передають «дефектний» ген своїм синам у спадок.
Історія хвороби Брутона офіційно розпочалася у 1952 р., коли її описав Огден Брутон – американський педіатр. На його ім'я надалі і було названо захворювання.
Почалося все із 8-річного хлопчика. Огден Брутон, обстежуючи його, дізнався, що за останні 4 роки він переніс 14 різних інфекційних захворювань. Серед них менінгіт, синусити, сепсис, отити і навіть пневмонія. Незвичайний випадок зацікавив педіатра, він направив пацієнта на дослідження. Вони показали, що сироватка крові не містить ніяких антитіл.
Понад 40 років знадобилося з'ясувати, як розвивається агаммаглобулінемія Брутона на молекулярному рівні. У 1993 р. двома групами вчених, незалежно одна від одної, було визначено причину хвороби – мутація у певному гені. Останній виявився нерецепторною тирозинкіназою. Викликає мутацію промутований білок, який є у гені.
Єдиною причиною захворювання є спадковість. Вона передається дитині за Х-зчепленим рецесивним типом і тільки при присутності в ДНК хромосом типу XY. Останнє буває у хлопчиків, тому захворювання діагностується тільки у них. Дівчаток воно не вражає, оскільки вони є володарями хромосоми ХХ ДНК.
Перші симптоми, що підтверджують присутність у дитини хвороби Брутона, видно вже у 3…6 міс. У них у крові спостерігається падіння кількості антитіл, які вони отримали у період розвитку в утробі від матері.
Надалі про агаммаглобулінемію у дитини починають свідчити часті інфекційні захворювання, які бувають і хронічними, і рецидивними. Для вродженої агаммаглобулінемії Брутона характерна така ознака – вони провокуються піогенними бактеріями. При дослідженнях виявляються пневмококи, гемофільна паличка, стафілококи та ін. Перелічені мікроорганізми здатні викликати розвиток гнійних запалень.
Для хвороби Брутона характерно те, що хлопчики, що захворіли, скаржаться на проблеми з ЛОР-органами. Крім цього можуть виникати проблеми зі шкірою (дерматит, екзема, піодермія), з жировою клітковиною під нею. Спостерігаються вони і в дихальних шляхах, і в шлунку, і кишечнику (наприклад, хронічна діарея). Буває, спостерігається кон'юнктивіт.
Постійні інфекційні захворювання, сповільнене зростання, характерні для хвороби Брутона, призводить до того, що хворі хлопчики виглядають менше фізично, ніж їхні однолітки.
У переліку інфекційних захворювань, на які можуть хворіти діти, які страждають на Х-зчеплену агаммаглобулінемію, синусит, енцефаліт, пневмонія, менінгіт, отит (частіше в середньому вусі). У таких хлопчиків найчастіше спостерігається алергія, аутоімунні захворювання. Вони значно частіше стикаються з онкологічними патологіями, з артритом, що вражає великі суглоби.
У переліку симптом можуть спостерігатися зменшені мигдалики та лімфатичні вузли. Іноді вони можуть бути взагалі відсутніми. Хвороба брутону характеризується так і тим, що при вакцинації хворої дитини від гепатиту В або поліомієліту призводить часто до більш швидкого розвитку всіх перелічених вище захворювань.
Діагностувати хворобу Брутона можна лише проводячи відповідні дослідження. Причому робити це потрібно в ранньому віці. Це дозволяє попередити надалі захворювання, пов'язані з вторинною інфекцією, зменшити ймовірність летального результату для хворих на агаммаглобулінемію.
Досліджують зазвичай кров. Хвороба імунодефіциту Брутона проявляється при цьому без гаммаглобуліну в протеїнограмі. До десяти разів може знижуватися рівень lg G, а lg А – у сотні разів. Фіксується при дослідженнях та значне зменшення В-лімфоцитів.
При діагностиці застосовують також рентген. Це дозволяє побачити відсутність мигдаликів або їх недорозвиненість. Рентгенограма фіксує зміни селезінки, патологію лімфовузлів. Після 5-річного віку хлопчики проводять бронхоскопію. Вона дозволяє виявляти можливі проблеми із дихальними шляхами. Використовують також ендоскопію, колоноскопію, щоб дослідити кишечник, шлунок і вчасно визначити зміни, що відбулися в них.
При агаммаглобулінемії призначається лише підтримуюча терапія. Полягає вона у воді хворому на препарати гамма-глобуліну. Його дози підбирають кожному пацієнту окремо. Загальним є те, що в результаті крові доза гамма-глобуліну повинна підтримуватися на рівні 3 г/л.
Крім того, протягом усього життя хворому потрібний прийом препаратів, які підтримують його імунітет. Усі терапевтичні заходи необхідно розпочинати у ранньому віці – 9…12 нед.
У разі інфекційних захворювань проводиться антибіотикотерапія, що дозволяє усувати симптоми хвороби Брутона.
Тут найкраще проводити заходи вже на етапі планування вагітності. Жінка піддається молекулярно-генетичній експертизі. Вона дозволяє виявити присутність у неї дефектного гена, що кодує нерецепторну тирозинкіназу.
Прогноз розвитку хвороби Брутона та її наслідків може бути позитивним. Це можливе лише за умови, якщо не порушується режим, встановлений лікарем для введення гамма-глобулінів, та своєчасно призначаються антибактеріальні ліки. Невиконання цього призводить, зазвичай, до різкого погіршення стану хворого. Може спостерігатись незворотна патологія, навіть смерть пацієнта.
В основі хвороби Брутона лежить спадковість, тому її профілактика марна. Парам, у яких таке захворювання є, планувати зачаття дитини потрібно лише після обстежень та консультацій у генетика.
Якщо ж батьки не знали про наявність у них дефектних генів, то дитина може з'явитися з агаммаглобулінемією. У разі профілактика зводиться до заходів, щоб захистити його від інфекцій.
Хвороба Брутона - це первинний гуморальний імунодефіцит, що виник через мутації в гені, який передається у спадок, внаслідок чого організм людини більш підданий різним інфекційним захворюванням, так як в організмі відбувається недостатнє виділення імунних молекул, так звані імуноглобуліни, які зобов'язані захищати організм від бактерій.
Американський педіатр Огден Брутон вперше зробив опис цього захворювання у 1952 році. Це був хлопчик, який страждає на хворобу Брутона, який був хворий на різні інфекційні захворювання. Десь з 4-х років він близько 14 разів хворів на пневмонію, лікувався від отиту, менінгіту, сепсису. При аналізі він не виявили антитіл. Група вчених у 1993 р. незалежно провели експеримент, в результаті якого було доведено, що Х-зчеплена хромосома виникла внаслідок мутації в гені нерецепторної тирозинкінази, згодом вона стала називатися тирозинкіназою Брутона.
Причини
Агаммаглобулінемія (хвороба Брутона) – досить рідкісна хвороба, на яку в основному страждають чоловіки, в поодиноких випадках це може бути жінка. Вона спровокована на генетичному рівні, ця хвороба стиснута з Х-хромосомою, внаслідок чого відбувається блокування росту абсолютно здорових імунних пре-В-клітин, так званих В-лімфоцитів. Це безпосередньо пов'язано з виникненням дефектатирозинкінази. Вона бере участь у трансдукції дозрівання В-лімфоцитів. Ген з дефектом знаходиться у межі хромосоми Хq21. Для того, щоб імуноглобуліни повноцінно захищали організм від різноманітних вірусів та бактерій, потрібно достатнє вироблення їх у крові. Але через цю хворобу вироблення імуноглобулінів уповільнюється або взагалі припиняється. Як правило, хвороба проявляється, коли дитині виповнюється понад півроку і вона має характер хронічного та рецидивуючого захворювання на бронхолегеневий апарат. Найчастіше виникають алергічні реакції на лікарські препарати.
У людей, які наражаються на цю хворобу, дуже високий ризик зараження такими бактеріями, як: гемофільна паличка, стрептококи, пневмококи. Найчастіше внаслідок супутніх інфекцій уражається шлунково-кишковий тракт, легені, шкіра, верхні дихальні шляхи, суглоби. Є велика ймовірність, що родичі хворого теж можуть зазнати цього захворювання, оскільки хвороба Брутона є спадковою.
Симптоми
Захворювання може супроводжуватися рядом наступних симптомів: хвороби верхніх дихальних шляхів, ураження шкіри, кон'юнктивіт (запалення очного яблука), бронхіт, пневмонія та ін. Найчастіше ці симптоми спостерігаються у дітей віком 4 років. Також можна відзначити в ряді симптомів бронхоектазію - розширення бронхів і напади астми, причому так. У період хвороби у хворих не відзначається збільшення лімфатичних вузлів, вони не страждають на гіперплазію мигдалин, аденоїдів. Агамаглобулінемія відбувається внаслідок мутації гена X-хромосоми, що кодує тирозинкіназу Брутона (ткБ, Btk – Brutontyrosinekinase). ТКБ займає дуже важливе значення у розвитку та дозріванні В-лімфоцитів. Антитіла і В-лімфоцити не зможуть сформуватися без ткБ, тому у хлопчиків можна помітити дуже маленькі мигдалики і лімфатичні вузли, що не розвиваються. Цій хворобі зазвичай схильні рецидивні гнійні інфекції легень, придаткових пазух носа, шкіри з інкапсульованими бактеріями (Streptococcus pneumoniae, Hemophilus influenzae), а також є велика ймовірність ураження центральної нервової системи, внаслідок вакцинації живої оральної поліомієліту. Зазвичай ці інфекції виникають у вигляді прогресуючого дерматоміозиту, який може супроводжуватися разом з енцефалітом або без нього.
Діагностика
Проводять діагностику методом проточної цитометрії, щоб виміряти кількість В-лімфоцитів, що циркулюють у крові. Проводиться сироватковий імуноелектрофорез, за допомогою нефелометрії вимірюється кількість імуноглобулінів, що містяться в крові.
Лікування
При лікуванні пацієнту вводять внутрішньовенно препарати імуноглобуліну 400 мг на 1 кг маси тіла для зміцнення та підтримки імунної системи в цілому, а також застосовують антибіотики, які гальмують та уповільнюють поширення та розвиток різних бактерій. Особливо важливе значення має своєчасне проведення антибіотикотерапії, якщо раптово відбувається прогресування інфекційного процесу, а із заміною антибіотиків бажано проводити лікування при бронхоектазії. При внутрішньовенному лікуванні досить покращується самопочуття хворих, які страждають на агаммаглобулінемію. Прогноз на одужання буде сприятливим, якщо на ранніх термінах хвороби призначити адекватне та відповідне лікування. Але якщо вчасно не розпочати лікування, існує велика ймовірність того, що тяжкі супутні хвороби можуть спричинити смерть хворого.
При спадковій гіпогаммаглобуленемії потрібна парентеральна антимікробна терапія. Для найкращого результату слід проводити її одночасно із супутньою чи замісною терапією. Термін лікування антибіотиками становить приблизно від 10-14 днів, але може зрости до 21 дня. Найпоширенішими антимікробними препаратами, що використовуються в лікуванні, є цефалоспорини, аміноглікозиди, сульфаніламіди та антибіотики пенніцилінового ряду.
З історії хвороби
Випадок, який був зафіксований у 1985 р. Народився малюк чоловічої статі з нормальною вагою 3500 г та зростом 53 см, пологи пройшли успішно без відхилень від норми. Мати, будучи вагітною, на 4 місяці перенесла ГРВІ. На першому місяці життя у хлопчика було помічено кон'юктивіт. Через 1 рік хлопчик стає постійним пацієнтом з діагнозом ГРЗ, бронхіт із задушливим кашлем, зі стабільними ентероколітами. У 2 роки дитина переносить пневмококовий менінгіт. А з генералізованими набряками він зіткнеться у віці 5 років, присутня і прискорена задишка, ціаноз. У нього з'являється біль у суглобах та в серці. Було обстежено печінку та селезінку та виявлено їх збільшення у розмірах у кілька разів, малюка терміново госпіталізували. При ретельному обстеженні в лабораторії було взято аналізи, в яких виявили виражену лімфоцитопенію, а також сліди імуноглобулінів усіх класів. Перед госпіталізацією було проведено лікування антибіотиками для усунення осередку інфекцій. З урахуванням цієї хвороби було застосовано внутрішньовенно імуноглобулін, у тому числі призначено антибіотикотерапію. Стан хворого покращився після проведення відповідного лікування, в організмі осередків інфекцій майже не залишилося. І через рік після хвороби пацієнта знову госпіталізували, але вже з двостороннім кон'юктивітом, а також бронхопневмонією. Повторно було призначено лікування внутрішньовенно гаммаглобуліном, одночасно з антибіотикотерапією. Після проведеного лікування пацієнта було виписано з такими рекомендаціями: постійний прийом гаммаглобуліну під ретельним контролем рівня крові. При цьому батьки хлопчика абсолютно здорові.
– це спадково обумовлене захворювання, при якому розвивається тяжкий первинний імунодефіцит (дефект імунного захисту організму) з вираженим зниженням рівня гамма-глобулінів у крові. Виявляється хвороба зазвичай у перші місяці та роки життя дитини, коли починають розвиватися повторні бактеріальні інфекції: отит, синусит, пневмонія, піодермія, менінгіт, сепсис. При обстеженні в периферичній крові та кістковому мозку практично відсутні сироваткові імуноглобуліни та B-клітини. Лікування агаммаглобулінемії полягає у довічній замісній терапії.
D80Імунодефіцити з переважною недостатністю антитіл

Агамаглобулінемія (спадкова гіпогамаглобулінемія, хвороба Брутона) – вроджений дефект гуморального імунітету, зумовлений мутаціями в геномі клітин, що призводить до недостатності синтезу B-лімфоцитів. В результаті порушується утворення імуноглобулінів усіх класів, та їх вміст у крові різко знижується аж до повної відсутності, розвивається первинний імунодефіцит. Низька реактивність імунної системи призводить до розвитку тяжких повторних гнійно-запальних захворювань ЛОР-органів, бронхів та легень, шлунково-кишкового тракту та мозкових оболонок. Хвороба Брутона зустрічається виключно у хлопчиків і спостерігається приблизно у 1-5 осіб із мільйона новонароджених, незалежно від раси та етнічної групи.
X-зчеплена форма спадкової агаммаглобулінемії виникає внаслідок пошкодження одного з генів X-хромосоми (розташований на Xq21.3-22.2). Цей ген відповідальний за синтез ферменту тирозинкінази, що бере участь у процесі утворення та диференціювання B-клітин. В результаті мутацій цього гена та блокування синтезу брутонівської тирозинкінази порушується формування гуморального імунітету. При агаммаглобулінемії молоді форми (пре-B-клітини) присутні в кістковому мозку, а їх подальше диференціювання та надходження у кровоносне русло порушено.
Відповідно, вироблення всіх класів імуноглобулінів практично не виробляється, і організм дитини стає беззахисним при проникненні хвороботворних бактерій (найчастіше це стрептококи, стафілококи та синьогнійна паличка). Подібний механізм порушень відзначається і у випадку з іншою формою спадкової агаммоглобулінемії – зчепленою з X-хромосомою та недостатністю гормону росту. Аутосомно-рецесивна форма розвивається внаслідок мутації кількох генів (µ-важких ланцюгів, гена λ5/14.1, гена адапторного білка та гена сигнальної молекули IgА).
Виділяють три форми спадкової агаммаглобулінемії:
Знижена реактивність гуморального імунітету при агаммаглобулінеміях призводить до розвитку повторних гнійно-запальних захворювань вже на першому році життя дитини (як правило, після припинення грудного вигодовування – 6-8 місяців). При цьому захисні антитіла від матері в організм дитини вже не надходять, а свої імуноглобуліни не виробляються. До 3-4 річного віку запальні процеси переходять у хронічну форму зі схильністю до генералізації. Гнійна інфекція при агаммаглобулінемії може вражати різні органи та системи.
З боку ЛОР-органів нерідко гнійні гайморити, етмоїдити, отити, причому гнійний отит частіше розвивається на першому році життя дитини, а синусити - в 3-5 років. З захворювань бронхолегеневої системи спостерігаються повторні бронхіти, пневмонії, абсцеси легені. Часто зустрічається ураження шлунково-кишкового тракту з наполегливою діареєю (проносами), спричиненою хронічним інфекційним ентероколітом (основні збудники – кампілобактерії, лямблії, ротавірус). На шкірних покривах виявляється імпетиго, мікробна екзема, рецидивуючий фурункульоз, абсцеси та флегмони.
Нерідким буває ураження очей (гнійні кон'юнктивіти), порожнини рота (виразкові стоматити, гінгівіти), кістково-м'язової системи (остеомієліти, гнійні артрити). Загалом клінічна картина агаммаглобулінемії характеризується поєднанням загальних симптомів, що спостерігаються при гнійній інфекції (висока температура тіла, озноб, біль у м'язах і головний біль, загальна слабкість, порушення сну та апетиту тощо) та ознак ураження конкретного органу (кашель, задишка) , утруднення носового дихання, гнійне відділення, діарея тощо).
Будь-яке інфекційне та соматичне захворювання у хворого на імунодефіцит протікає важко, тривало і супроводжується ускладненнями. Тяжкий перебіг агаммоглобулінемії може ускладнюватися розвитком менінгіту, вірусного енцефаломієліту, поствакцинального паралітичного поліомієліту, сепсису. На фоні захворювання підвищено ймовірність розвитку аутоімунних та онкологічних захворювань. Загибель пацієнтів часто настає від інфекційно-токсичного шоку.
При клінічному огляді пацієнта лікарем алергологом-імунологом виявляються ознаки гнійно-запального ураження того чи іншого органу (тканини) та симптоми, що підтверджують знижену реактивність імунної системи: гіпоплазія мигдаликів, зменшення периферичних лімфатичних вузлів. Виражено і ознаки відставання у фізичному розвитку дитини.
Лабораторне дослідження крові виявляє виражене зниження рівня імуноглобулінів в імунограмі (IgA та IgM< 20 мг/дл – снижение в сто раз, IgG < 200 мг/дл – в десять раз) или их полное отсутствие. В периферической крови обнаруживается глубокий дефицит B-клеток (менее 1%), в костном мозге практически отсутствуют плазмоциты. Что касается клеточного иммунитета, то содержание T-лимфоцитов может быть в норме. При гистологическом исследовании лимфоидной ткани герминативные (зародышевые) центры и плазматические клетки отсутствуют.
Диференціальна діагностика спадкової агаммаглобулінемії проводиться з іншими первинними та вторинними імунодефіцитними станами (генетичними порушеннями, ВІЛ та цитомегаловірусною інфекцією, вродженою краснухою та токсоплазмозом, злоякісними новоутвореннями та системними порушеннями.).
Необхідна довічна замісна терапія антитіловмісними препаратами. Зазвичай використовується введення внутрішньовенного імуноглобуліну, а за його відсутності нативної плазми від здорових постійних донорів. При вперше встановленому діагнозі агаммаглобулінемії замісне лікування проводиться в режимі насичення до досягнення рівня імуноглобуліну IgG вище 400 мг/дл, після чого при відсутності активного гнійно-запального процесу в органах і тканинах можна переходити до підтримуючої терапії з введенням профілактичних доз.
Будь-який епізод бактеріальної гнійної інфекції, незалежно від локалізації запального процесу, вимагає проведення адекватної антибактеріальної терапії, яка виконується одночасно із замісним лікуванням. Найчастіше при агаммаглобулінемії використовуються антибактеріальні засоби групи цефалоспоринів, аміноглікозидів, макролідів, а також антибіотики пеніцилінового ряду. Тривалість лікування при цьому у кілька разів перевищує стандартну при цьому захворюванні.
Симптоматичне лікування проводиться з урахуванням конкретного ураження того чи іншого органу (промивання навколоносових пазух носа антисептиками, виконання вібраційного масажу грудної клітки та постурального дренажу при бронхітах та пневмоніях тощо).
Якщо агаммаглобулінемія виявлена в ранньому віці до настання важких ускладнень, і адекватна стану пацієнта замісна терапія розпочато своєчасно, можливе збереження нормального життя протягом багатьох років. Однак у більшості випадків діагностика спадкових порушень гуморального імунітету здійснюється надто пізно, коли вже розвинулися незворотні хронічні гнійно-запальні захворювання органів та систем організму. І тут прогноз при агаммаглобулинемии несприятливий.

У 2018 році Різдвяний пост розпочнеться 28 листопада. У цей період православні віруючі готуються до зустрічі Різдва.

Створення сім'ї – мрія більшості жінок. Вони хочуть мати люблячого чоловіка і купу дітей. Тільки от не завжди стосунки...

Ця стаття містить: найсильніша молитва від розлучення - інформація взята зі всіх куточків світла, електронної мережі та...

Інформаційний сайт про ікони, молитви, православні традиції. Молитва від скандалів та сварок у сім'ї, з чоловіком, з дітьми...

Який же Новий рік без шампанського, мандарин, «Олів'є», заливного та всіма улюбленої «Оселедця під шубою». Ось із останнім...

Підготуємо необхідні інгредієнти для печива. Перше, що потрібно зробити - це поставити воду кип'ятитися. Нам необхідний...

Чи можна оформити працівника посаду фінансовий директор - головний бухгалтер. Головний бухгалтер стверджує, що...

Керівник невеликого бізнесу може вести бюджет самостійно. ПЕРЕВІРЕНО! Якщо займатися веденням...

Створення нових проектів передбачає попереднє економічне обґрунтування їхньої доцільності, наступне...
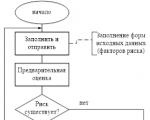
Звітність формується РМ, проходить узгодження (схвалення) Комітету з ризиків за Правління та передається на...

На краю великого луга, на довгій смарагдовій травинці жила крихітна Божа Корівка. Маленька Божа...

Нині досить поширене звернення людини до зірок. За допомогою гороскопу людина може дізнатися...

Діловий чи приятельський. Якщо ж переслідували незнайомку - значить рівень вашої довіри до жіночої статі.

по соннику ФрейдаЯкщо вам снилося, як ви ловили рибу, значить, у реальному житті ви насилу можете відключитися...

Створення сім'ї – мрія більшості жінок. Вони хочуть мати люблячого чоловіка і купу дітей. Тільки от не завжди...

Ця стаття містить: найсильніша молитва від розлучення - інформація взята зі всіх куточків світла, електронної...