Що можна і не можна на Різдвяний піст?
У 2018 році Різдвяний пост розпочнеться 28 листопада. У цей період православні віруючі готуються до зустрічі Різдва.
Процес створення нових лікарських засобів виконується відповідно до міжнародних стандартів GLP (Good Laboratory Practice Якісна лабораторна практика), GMP (Good Manufacturing Practice Якісна виробнича практика) та GCP (Good Clinical Practice Якісна клінічна практика).
Клінічні випробування лікарських засобів включають систематичне вивчення досліджуваного препарату на людях для перевірки його лікувальної дії або виявлення небажаної реакції, а також вивчення всмоктування, розподілу, метаболізму та виведення з організму для визначення його ефективності та безпеки.
Клінічні дослідження лікарського засобу є необхідним етапом розробки будь-якого нового препарату або розширення показань для застосування лікарського засобу, вже відомого лікарям. На початкових етапах розробки лікарських засобів проводяться хімічні, фізичні, біологічні, мікробіологічні, фармакологічні, токсикологічні та інші дослідження на тканинах (in vitro) чи лабораторних тварин. Це так звані доклінічні дослідження, метою яких є одержання науковими методами оцінок та доказів ефективності та безпеки лікарських засобів. Однак ці дослідження не можуть дати достовірної інформації про те, як препарати, що вивчаються, діятимуть у людини, тому що організм лабораторних тварин відрізняється від людського і за фармакокінетичними характеристиками і по реакції органів і систем на ліки. Тому необхідно проведення клінічних випробувань лікарських засобів у людини.
Клінічне дослідження (випробування) лікарського засобу - це системне вивчення лікарського препарату за допомогою застосування його у людини (пацієнта або здорового добровольця) з метою оцінки його безпеки та ефективності, а також виявлення або підтвердження його клінічних, фармакологічних, фармакодинамічних властивостей, оцінки всмоктування, розподілу, метаболізму, виведення та взаємодії з іншими лікарськими засобами. Рішення про початок клінічного дослідження приймає замовник, який відповідає за організацію, контроль та фінансування дослідження. Відповідальність за практичне проведення дослідження покладено на дослідника. Як правило, спонсором є фармацевтичні компанії - розробники лікарських засобів, однак у ролі спонсора може виступати і дослідник, якщо дослідження розпочато з його ініціативи та він несе повну відповідальність за його проведення.
Клінічні дослідження повинні проводитися відповідно до основоположних етичних принципів Гельсінської Декларації, Правил GSP (Good Clinical Practice, Належна Клінічна Практика) та чинних нормативних вимог. До початку клінічного дослідження має бути проведена оцінка співвідношення передбачуваного ризику з очікуваною користю для випробуваного та суспільства. На чільне місце ставиться принцип пріоритету прав, безпеки і здоров'я випробуваного над інтересами науки і суспільства. Випробуваний може бути включений у дослідження лише на підставі добровільної інформованої згоди (ІВ), одержаної після детального ознайомлення з матеріалами дослідження. Пацієнти (добровольці), які беруть участь у випробуванні нового препарату, повинні отримати інформацію про сутність та можливі наслідки випробувань, очікувану ефективність ліків, рівень ризику, укласти договір про страхування життя та здоров'я в порядку, передбаченому законодавством, а під час випробувань перебувати під постійним наглядом кваліфікованого персоналу. У разі виникнення загрози здоров'ю чи життю пацієнта, а також за бажанням пацієнта або його законного представника керівник клінічних випробувань зобов'язаний призупинити випробування. Крім того, клінічні випробування зупиняються у разі відсутності чи недостатньої ефективності ліків, а також порушення етичних норм.
Перший етап клінічних випробувань ЛЗ здійснюється на 30 – 50 добровольцях. Наступний етап - розширені випробування на базі 2 - 5 клінік із залученням великої кількості (кілька тисяч) хворих. У цьому заповнюються індивідуальні карти хворих із докладним описом результатів різних досліджень - аналізів крові, сечі, УЗД та інших.
Кожен лікарський засіб проходить 4 фази (етапу) клінічних досліджень.
Фаза I. Перший досвід застосування нової активної речовини у людини. Найчастіше дослідження починаються у добровольців (дорослі здорові чоловіки). Головна мета досліджень – вирішити, чи варто продовжувати роботу над новим препаратом, і якщо вдасться встановити дози, які будуть використовуватися у пацієнтів під час II фази клінічних досліджень. У ході цієї фази дослідники отримують попередні дані про безпеку нового препарату та вперше описують його фармакокінетику та фармакодинаміку у людини. Іноді неможливо провести дослідження І фази у здорових добровольців через токсичність цього препарату (лікування онкологічних захворювань, СНІДу). У цьому випадку проводяться нетерапевтичні дослідження за участю пацієнтів із цією патологією у спеціалізованих установах.
Фаза ІІ. Зазвичай це перший досвід застосування у пацієнтів із захворюванням, для лікування якого передбачається використовувати препарат. Друга фаза ділиться на IIa та IIb. Фаза IIa - це терапевтичні пілотні дослідження (pilot studies), оскільки отримані результати забезпечують оптимальне планування подальших досліджень. Фаза IIb - це більш широкі дослідження у пацієнтів із захворюванням, яке є основним показанням до призначення нового лікарського засобу. Головна мета – довести ефективність та безпеку препарату. Результати цих досліджень (pivotal trial) є основою для планування досліджень III фази.
Фаза ІІІ. Багатоцентрові випробування за участю великих (і наскільки можна, різноманітних) груп пацієнтів (загалом 1000-3000 людина). Основна мета - отримання додаткових даних про безпеку та ефективність різних форм препарату, про характер найчастіших небажаних реакцій тощо. Найчастіше клінічні дослідження цієї фази – подвійні сліпі контрольовані, рандомізовані, а умови досліджень максимально наближені до звичайної реальної рутинної медичної практики. Дані, отримані у клінічних дослідженнях III фази, є основою для створення інструкцій щодо застосування препарату та для вирішення його реєстрації Фармакологічним комітетом. Рекомендація до клінічного застосування в медичній практиці вважається обґрунтованою, якщо новий препарат:
Фаза ІV. Дослідження проводяться після початку продажу препарату з метою отримати більш детальну інформацію про тривале застосування у різних групах пацієнтів та за різних факторів ризику тощо. і таким чином повніше оцінити стратегію застосування лікарського засобу. У дослідженні бере участь велика кількість пацієнтів, це дозволяє виявити раніше невідомі і небажані явища, що рідко зустрічаються.
Якщо лікарський засіб збираються застосовувати за новим показанням, ще не зареєстрованим, для цього проводяться додаткові дослідження, починаючи з фази II. Найчастіше практично проводять відкрите дослідження, у якому лікаря і хворому відомий спосіб лікування (досліджуваний препарат чи препарат порівняння).
При випробуванні простим сліпим методом хворий не знає, який препарат він приймає (це може бути плацебо), а при використанні подвійного сліпого методу про це не обізнані ні хворий, ні лікар, а лише керівник випробування (у сучасному клінічному дослідженні нового лікарського засобу беруть участь чотири сторони: спонсор дослідження (найчастіше це фармацевтична компанія-виробник), монітор – контрактна дослідницька організація, лікар-дослідник, пацієнт). Крім того, можливі потрійні сліпі дослідження, коли ні лікар, ні пацієнт, ні ті, хто організує дослідження та обробляє його дані, не знають призначеного лікування у конкретного пацієнта.
Якщо лікарі знатимуть, який пацієнт лікується якимсь засобом, вони мимоволі можуть давати оцінки лікуванню залежно від своїх переваг чи пояснень. Застосування сліпих методів підвищує достовірність результатів клінічного випробування, усуваючи вплив суб'єктивних чинників. Якщо хворий знає, що він отримує нові багатообіцяючі ліки, то ефект лікування може бути пов'язаний з його заспокоєнням, задоволеністю тим, що досягнуто найбажанішого лікування з можливих.
Плацебо (лат. placere - подобатися, цінуватися) позначає препарат, який явно не має ніяких цілющих властивостей. Великий енциклопедичний словник визначає плацебо як «лікарську форму, що містить нейтральні речовини. Застосовують для вивчення ролі навіювання в лікувальному ефекті будь-якої лікарської речовини як контроль при дослідженні ефективності нових лікарських препаратів». випробування якість ліки препарат
Негативні плацебо-ефекти звуться ноцебо. Якщо пацієнт знає, які побічні дії є у препарату, то у 77% випадків вони виникають у нього, коли він приймає плацебо. Віра у той чи інший ефект може зумовити появу побічної дії. Згідно з коментарем Всесвітньої медичної асоціації до статті 29 Гельсінкської декларації, «…застосування плацебо виправдане, якщо це не призведе до підвищення ризику заподіяння серйозної чи незворотної шкоди здоров'ю…», тобто якщо хворий не залишиться без ефективного лікування.
Існує термін «повні сліпі дослідження», коли всі сторони дослідження не мають інформації про тип лікування конкретного хворого до завершення аналізу отриманих результатів.
Рандомізовані контрольовані випробування є стандартом якості наукових досліджень ефективності лікування. Для дослідження спочатку відбираються пацієнти з великої кількості людей з станом, що вивчається. Потім цих пацієнтів поділяють випадково на дві групи, порівняні за основними прогностичними ознаками. Групи формуються випадковим чином (рандомізація) шляхом використання таблиць випадкових чисел, у яких кожна цифра чи комбінація цифр має рівну ймовірність відбору. Це означає, що пацієнти однієї групи будуть в середньому мати ті ж характеристики, що і пацієнти іншої. Крім того, до проведення рандомізації слід переконатися, що характеристики захворювання, про які відомо, що вони сильно впливають на результат, зустрічаються в експериментальних та контрольних групах з однаковою частотою. Для цього треба спочатку розподілити пацієнтів за підгрупами з однаковим прогнозом і лише потім рандомізувати їх окремо у кожній підгрупі – стратифікована рандомізація. Експериментальна група (група лікування) піддається втручанню, яке, як очікується, буде корисним. Контрольна група (група порівняння) знаходиться в таких самих умовах, як і перша, за винятком того, що її пацієнти не піддаються досліджуваному втручанню.
ГОСТ Р 56701-2015
НАЦІОНАЛЬНИЙ СТАНДАРТ РОСІЙСЬКОЇ ФЕДЕРАЦІЇ
ЛІКИ ЗАСОБИ ДЛЯ МЕДИЧНОГО ЗАСТОСУВАННЯ
Посібник з планування доклінічних досліджень безпеки з метою подальшого проведення клінічних досліджень та реєстрації лікарських засобів
Medicines for medical applications. Guidance on nonclinical safety studies for conduct of human clinic trials and marketing authorization for pharmaceuticals
ГКС 11.020
11.120.01
Дата введення 2016-07-01
Передмова
1 ПІДГОТОВЛЕНО Технічним комітетом зі стандартизації ТК 458 "Розробка, виробництво та контроль якості лікарських засобів" на основі власного автентичного перекладу російською мовою документа, зазначеного у пункті 4
2 ВНЕСЕН Технічним комітетом зі стандартизації ТК 458 "Розробка, виробництво та контроль якості лікарських засобів"
3 ЗАТВЕРДЖЕНИЙ І ВВЕДЕНИЙ У ДІЮ Наказом Федерального агентства з технічного регулювання та метрології від 11 листопада 2015 р. N 1762-ст.
4 Цей стандарт ідентичний міжнародному документу ICH M3(R2):2009* "Посібник з планування доклінічних досліджень безпеки з метою подальшого проведення клінічних досліджень та реєстрації лікарських засобів" (ICH M3(R2):2009 "Guidance on nonclinical safety studies for the conduct of human clinic trials and marketing authorization for pharmaceuticals"). Найменування цього стандарту змінено щодо найменування зазначеного міжнародного документа для ув'язування із найменуваннями, прийнятими у існуючому комплексі стандартів "Лікарські засоби для медичного застосування". При застосуванні цього стандарту рекомендується використовувати замість посилальних міжнародних стандартів відповідні їм національні стандарти Російської Федерації, зазначені у додатку ТАК
________________
* Доступ до міжнародних та зарубіжних документів, згаданих у тексті, можна отримати, звернувшись до Служби підтримки користувачів . - Примітка виробника бази даних.
5 ВВЕДЕНО ВПЕРШЕ
Правила застосування цього стандарту встановлені вГОСТ Р 1.0-2012 (Розділ 8). Інформація про зміни до цього стандарту публікується у щорічному (станом на 1 січня поточного року) інформаційному покажчику "Національні стандарти", а офіційний текст змін та поправок - у інформаційному покажчику "Національні стандарти", який щомісяця видається. У разі перегляду (заміни) або скасування цього стандарту відповідне повідомлення буде опубліковано у найближчому випуску щомісячного інформаційного покажчика "Національні стандарти". Відповідна інформація, повідомлення та тексти розміщуються також в інформаційній системі загального користування – на офіційному сайті Федерального агентства з технічного регулювання та метрології в мережі Інтернет (www.gost.ru)
Вступ
Метою цього стандарту є встановлення єдиних з країнами Європейського союзу, Сполученими Штатами Америки, Японією та іншими країнами, що застосовують міжнародні керівництва ICH, підходів до планування доклінічних досліджень лікарських засобів для обґрунтування можливості проведення клінічних досліджень певного характеру та тривалості, а також подальшої державної реєстрації.
Стандарт сприяє своєчасному проведенню клінічних досліджень, зменшенню використання лабораторних тварин відповідно до принципу 3R (reduce/refine/replace, скорочення/покращення/заміна) та скорочення використання інших ресурсів при розробці лікарських препаратів. Слід розглядати можливість використання нових альтернативних методів in vitroдля оцінки безпеки. Дані методи при їх належній валідації та прийнятті всіма регуляторними органами країн, що застосовують керівництво ICH, можуть використовуватися замість існуючих стандартних методів.
Цей стандарт сприяє проведенню безпечної етичної розробки лікарських препаратів та їх доступності для пацієнтів.
Доклінічна оцінка безпеки, що проводиться з метою державної реєстрації лікарських засобів, зазвичай включає наступні етапи: фармакологічні дослідження, загальнотоксикологічні дослідження, токсикокінетичні та доклінічні фармакокінетичні дослідження, вивчення репродуктивної токсичності, дослідження генотоксичності. Для лікарських препаратів, які мають певні властивості або призначені для тривалого застосування, необхідно також проведення оцінки канцерогенного потенціалу. Необхідність проведення інших доклінічних досліджень з метою оцінки фототоксичності, імунотоксичності, токсичності на нестатевозрілих тваринах та виникнення лікарської залежності визначається в індивідуальному порядку. Цей стандарт визначає необхідність проведення доклінічних досліджень та їх взаємозв'язок з подальшими клінічними дослідженнями у людини.
На даний момент у країнах, що використовують керівництво ICH, отримано суттєві досягнення у гармонізації термінів доклінічних досліджень безпеки для проведення клінічних досліджень лікарських засобів, описаних у цьому стандарті. Однак у деяких сферах відмінності зберігаються. Регуляторні органи та виробники продовжують розгляд цих відмінностей та працюють з метою подальшого вдосконалення процесу розробки лікарських препаратів.
Цей стандарт встановлює рекомендації щодо планування доклінічних досліджень безпеки з метою подальшого проведення клінічних досліджень та реєстрації лікарських засобів.
Цей стандарт застосовується у всіх випадках розробки лікарських препаратів і є загальними положеннями щодо їх розробки.
Для лікарських препаратів, отриманих із застосуванням біотехнологічних методів, відповідні дослідження безпеки необхідно проводити згідно з керівництвом ICH S6 з доклінічним дослідженням біотехнологічних препаратів. Для цих лікарських засобів цей стандарт застосовується лише щодо черговості проведення доклінічних досліджень залежно від фази клінічної розробки.
Для оптимізації та прискорення розробки лікарських препаратів, призначених для лікування загрозливих для життя або тяжких захворювань (наприклад, поширений рак, стійка ВІЛ-інфекція, стани, зумовлені вродженою ферментативною недостатністю), ефективна терапія яких зараз відсутня, також застосовують індивідуальний підхід як до токсикологічної оцінки, і до клінічної розробки. У цих випадках, а також щодо лікарських препаратів на основі інноваційних терапевтичних речовин (наприклад, мала інтерферуюча РНК) та ад'ювантів вакцин, певні дослідження можуть бути скорочені, змінені, додані або виключені. За наявності посібників ICH щодо окремих фармакотерапевтичних груп лікарських препаратів необхідно керуватися останніми.
Розробка лікарського препарату є поетапним процесом, що включає оцінку даних про його ефективність і безпеку як для тварин, так і для людини. Основні цілі доклінічної оцінки безпеки лікарського препарату включають визначення токсичного впливу на органи-мішені, його залежності від дози, зв'язку з експозицією (системним впливом), а також, якщо застосовно, потенційної оборотності токсичних ефектів. Ці дані використовують для визначення вихідної безпечної дози та діапазону доз для клінічних досліджень, а також для встановлення параметрів клінічного моніторингу потенційних небажаних ефектів. Доклінічні дослідження безпеки, незважаючи на їх обмежений характер на початку клінічної розробки, мають бути достатніми для вказівки потенційних небажаних ефектів, які можуть виникнути в умовах запланованих клінічних досліджень.
Клінічні дослідження проводяться для вивчення ефективності та безпеки лікарського препарату, починаючи з відносно низької системної експозиції у невеликої кількості суб'єктів. У подальших клінічних дослідженнях експозиція лікарського препарату збільшується шляхом збільшення тривалості застосування та (або) розміру популяції пацієнтів, які беруть участь у дослідженні. Клінічні дослідження слід розширювати при належному підтвердженні безпеки за результатами раніше проведених клінічних досліджень та на підставі додаткових доклінічних даних щодо безпеки, які отримують у міру просування клінічної розробки.
Клінічні або доклінічні дані щодо серйозних небажаних ефектів можуть вплинути на продовження клінічних досліджень. У рамках загального плану клінічної розробки ці дані необхідно розглядати для визначення доцільності проведення та дизайну додаткових доклінічних та (або) клінічних досліджень.
Клінічні дослідження проводяться за фазами, які мають різні назви у різних країнах. У цьому стандарті використовується термінологія, що застосовується в посібнику ICH Е8 за загальними принципами проведення клінічних досліджень лікарських препаратів. Однак, оскільки спостерігається стійка тенденція до об'єднання фаз клінічної розробки, у зазначеному документі в деяких випадках також визначено взаємозв'язок доклінічних досліджень з тривалістю та масштабом клінічних досліджень, а також характеристиками суб'єктів, що беруть у них участь (цільової популяції).
Планування та дизайн доклінічних досліджень безпеки та клінічних досліджень у людини повинні ґрунтуватися на науковому підході та відповідати етичним принципам.
2.1 Вибір високих доз для дослідження загальнотоксичної дії
Потенційні клінічно значущі ефекти в токсикологічних дослідженнях, як правило, можуть бути повноцінно вивчені при введенні доз, близьких до дози, що максимально переноситься (МПД, MTD). Однак не потрібно підтверджувати MTD у кожному дослідженні. Також допускається використовувати обмежено високі дози, включаючи дози, кратні дозам, які планується застосовувати в клінічній практиці (клінічна експозиція) або за яких досягається максимально досяжна експозиція (експозиція насичення), або прийнятні максимальні дози (MFD). Використання цих обмежено високих доз (докладний опис наведено далі і на малюнку 1) дозволяє виключити введення тварин доз, які не представляють додаткової інформації для прогнозування клінічної безпеки. Описаний підхід узгоджується з аналогічними рекомендаціями щодо дизайну досліджень репродуктивної токсичності та канцерогенності, в яких вже визначено обмежено високі дози та (або) експозиції.
Обмежено висока доза 1000 мг/кг/добу для досліджень гострої, субхронічної та хронічної токсичності для гризунів та негризунів вважається придатною для всіх випадків, крім обговорюваних нижче. У деяких випадках, коли доза 1000 мг/кг/добу не забезпечує 10-кратного перевищення клінічної експозиції, а клінічна доза препарату перевищує 1 г/добу, тоді дози в токсикологічних дослідженнях слід обмежити 10-кратною дозою для досягнення клінічної експозиції, дозою 2000 мг /кг/добу або використовувати MFD, вибравши найменшу. У окремих випадках, коли доза 2000 мг/кг/добу нижче клінічної експозиції, може використовуватися більш висока доза аж до MFD.
Дози, що забезпечують 50-кратне перевищення системної експозиції (зазвичай визначається за груповим середнім значенням AUC (примітка 1) вихідної речовини або фармакологічно активної молекули проліки) у порівнянні з системною клінічною експозицією, також вважаються прийнятними як максимальні дози для досліджень гострої токсичності та токсичності при багаторазове введення у будь-яких видів тварин.
Для початку III фази клінічних досліджень США токсикологічні дослідження з використанням обмежено високих доз проводяться щонайменше одному вигляді тварин із дозою, що забезпечує 50-кратную експозицію. Якщо цей підхід не застосовується, рекомендується проведення дослідження на одному виді тварин протягом 1 місяця і більше з використанням обмежено високої дози 1000 мг/кг, MFD або MTD, вибравши найменшу. Однак в окремих випадках таке дослідження може не знадобитися, якщо у дослідженні меншої тривалості токсична дія спостерігалася при використанні доз, що перевищують дози 50-кратної експозиції. Якщо до дослідження загальної токсичності включаються кінцеві точки з оцінки генотоксичності, тоді потрібна максимальна доза повинна підбиратися на підставі MFD, MTD або обмежено високої дози 1000 мг/кг/добу.
Примітка 1 - У цьому документі під "експозицією" загалом мається на увазі середнє значення AUC у групі. У деяких випадках (наприклад, якщо сполука або клас сполук здатні викликати гострі серцево-судинні зміни або симптоми пов'язані з впливом на ЦНС) більш доцільним є визначення меж експозиції за середніми значеннями С у групах.
Визначення фармакологічних та фармакодинамічних досліджень безпеки наведено у Посібнику ICH S7A.
Основний набір досліджень фармакологічної безпеки включає оцінку впливу на серцево-судинну, центральну нервову та дихальну системи. В цілому ці дослідження повинні бути проведені до початку клінічної розробки відповідно до принципів, викладених у посібниках ICH S7A та S7B з вивчення фармакологічної безпеки лікарських препаратів та доклінічної оцінки здатності лікарських препаратів для медичного застосування уповільнювати реполяризацію шлуночків (подовжувати інтервал QT). При необхідності додаткові та подальші дослідження фармакологічної безпеки можуть бути проведені на пізніх стадіях клінічної розробки. З метою скорочення практики використання лабораторних тварин слід по можливості включати до протоколів загальнотоксичних досліджень інші оцінки in vivoяк додаткові.
Метою первинних фармакодинамічних досліджень ( in vivoта (або) in vitro) є встановлення механізму дії та (або) фармакологічних ефектів діючої речовини у взаємозв'язку з його пропонованим терапевтичним застосуванням. Такі дослідження, як правило, проводяться на початковому етапі фармацевтичної розробки і, як правило, проводяться не відповідно до Принципів належної лабораторної практики (GLP). Результати цих досліджень можуть бути використані при виборі дози як для доклінічних, так і для клінічних досліджень.
До початку клінічних досліджень, як правило, слід оцінити метаболічний профіль та ступінь зв'язування з білками плазми тварин та людини in vitro, а також дані про системну експозицію (Керівництво ICH S3A з токсикокінетичних досліджень) у видів тварин, що використовуються в токсикологічних дослідженнях з багаторазовим введенням. До початку клінічних досліджень у великої кількості випробуваних або протягом тривалого періоду часу (як правило, до початку фази III клінічних досліджень) повинні бути отримані дані про фармакокінетику (ФК) (тобто відомості про абсорбцію, розподіл, метаболізм та виведення) у досліджуваних видів тварин та біохімічні дані, одержувані in vitro, значимі виявлення потенційних лікарських взаємодій. Ці дані використовуються для порівняння метаболітів у людини та тварин та визначення необхідності проведення додаткових досліджень.
Доклінічна характеристика властивостей метаболіту у людини необхідна тільки тоді, коли його експозиція перевищує 10% від сукупної експозиції лікарського препарату і величина експозиції у людини значно перевищує таку, що спостерігалася в токсикологічних дослідженнях. Такі дослідження мають бути проведені для отримання дозволу на проведення ІІІ фази клінічних досліджень. Для лікарських препаратів, добова доза яких вводиться не перевищує 10 мг, такі дослідження можуть знадобитися при більш високих частках метаболітів. Деякі метаболіти не є об'єктами токсикологічних досліджень (наприклад, більшість кон'югатів з метіоніном) і не вимагають вивчення. Необхідність доклінічного вивчення метаболітів, які можуть мати можливі токсикологічні ефекти (наприклад, властивий тільки людині метаболіт), необхідно розглядати в індивідуальному порядку.
Традиційно дані про гостру токсичність отримують із досліджень токсичності при одноразовому введенні препарату на двох видах ссавців з використанням запропонованого для клінічного застосування та парентеральних способів введення. Однак цю інформацію також можна отримати з належним чином проведених досліджень з ескалацією доз або короткострокових досліджень з використанням діапазону доз, в яких визначають MTD для тварин, які використовуються для вивчення загальнотоксичної дії.
У тих випадках, коли інформацію про гостру токсичність можна отримати з інших досліджень, окремі дослідження з одноразовим введенням препарату проводити не рекомендується. Дослідження, що забезпечують отримання відомостей про гостру токсичність, можуть бути обмежені використанням тільки шляхом введення, запропонованим для клінічного застосування, і можуть бути проведені не відповідно до вимог GLP, якщо в дослідженнях токсичності з багаторазовим введенням, проведених відповідно до вимог GLP, використовувався шлях введення препарату, що пропонується для клінічного використання. Летальність не повинна бути обов'язковою кінцевою точкою у дослідженнях гострої токсичності. У деяких особливих випадках (наприклад, при дослідженні мікродоз див. розділ 7) дослідження гострої токсичності або дослідження з одноразовим введенням дози можуть бути головним обґрунтуванням можливості проведення клінічних досліджень у людини. У цих випадках вибір високої дози може відрізнятися від описаного в розділі 1.1, але повинен враховувати передбачувану клінічну дозу та шлях введення препарату. Ці дослідження необхідно проводити відповідно до вимог GLP.
Інформація про гостру токсичність лікарських препаратів може використовуватися для прогнозування наслідків передозування у людини, вона має бути доступна перед початком клінічних досліджень ІІІ фази. Більш рання оцінка гострої токсичності може знадобитися для препаратів, пропонованих для терапії груп пацієнтів, які мають високий ризик передозування (наприклад, депресія, біль, деменція) у клінічних дослідженнях, що проводяться амбулаторно.
Рекомендована тривалість досліджень токсичності з багаторазовим введенням залежить від тривалості, показань до застосування та спрямованості запланованого подальшого клінічного дослідження. Як правило, тривалість досліджень токсичності на тваринах, які проводяться на двох видах тварин (один з яких негризуни), повинна бути рівною або перевищувати заплановану тривалість клінічних досліджень аж до рекомендованої максимальної тривалості досліджень токсичності з багаторазовим введенням (таблиця 1). Обмежені високі дози/експозиції, які вважаються придатними для досліджень токсичності при багаторазовому введенні, описані у 2.1.
У тих випадках, коли в клінічних дослідженнях спостерігається значний терапевтичний ефект, їх тривалість в індивідуальному порядку може бути збільшена порівняно з тривалістю досліджень токсичності з багаторазовим введенням, використаним як основи для проведення клінічних досліджень.
6.1 Дослідження, необхідні для проведення клінічної розробки
Як правило, для обґрунтування можливості будь-яких клінічних досліджень тривалістю до двох тижнів достатньо проведення дослідження токсичності з багаторазовим введенням на двох видах (один з яких негризуни) з мінімальною тривалістю два тижні (таблиця 1). Для обґрунтування клінічних досліджень більшої тривалості потрібні дослідження токсичності щонайменше такої ж тривалості. Для обґрунтування клінічних досліджень із тривалістю понад 6 місяців потрібне проведення 6-місячного дослідження на гризунах та 9-місячного дослідження на негризунах (винятки зазначені у примітках до таблиці 1).
Таблиця 1 – Рекомендована тривалість токсикологічних досліджень з багаторазовим введенням, необхідних для обґрунтування проведення клінічних випробувань
Максимальна тривалість клінічного дослідження | ||
Гризуни | Негризуни |
|
До двох тижнів | 2 тижні | |
Від двох тижнів до шести місяців | Така сама, як у клінічних дослідженнях |
|
Понад шість місяців | 6 місяців | 9 місяців |
У США для обґрунтування клінічних досліджень з одноразовим введенням дози лікарського препарату допускається як альтернатива 2-тижневим дослідженням використання розширеного вивчення токсичності з одноразовим введенням дози препарату (див. примітку "с" до таблиці 3). Клінічні дослідження тривалістю менше 14 днів можуть бути обґрунтовані дослідженнями токсичності такої самої тривалості. У деяких випадках клінічні дослідження з тривалістю більшою, ніж 3 місяці, можуть бути розпочаті за наявності результатів 3-місячних досліджень на гризунах та негризунах за умови, що результати завершених досліджень хронічної токсичності на гризунах та негризунах відповідно до національних регуляторних вимог до клінічних досліджень можуть бути представлені до перевищення клінічного застосування лікарського препарату понад 3 місяці. Для серйозних або загрозливих для життя захворювань або в індивідуальному порядку таке продовження можливе за наявності результатів повністю завершених досліджень хронічної токсичності на гризунах та результатів прижиттєвих досліджень та даних некропсії у дослідженнях на негризунах. Повні патогістологічні дані негризунів повинні бути отримані протягом наступних 3 місяців. Можливі випадки, коли препарат призначений для педіатричного застосування, а наявні доклінічні дослідження на тваринах (токсикологічні чи фармакологічні) вказують на потенційний вплив на процеси розвитку органів-мішеней. У цих випадках можуть знадобитися довгострокові дослідження токсичності, розпочаті на нестатевозрілих тваринах (див. розділ 12). У Європейському союзі токсикологічні дослідження протягом шести місяців на негризунах вважаються достатніми. Однак, якщо проведено дослідження більшої тривалості, проведення додаткових досліджень протягом 6 місяців неприйнятне. Далі наведено приклади, коли дослідження на негризунах 6-місячної тривалості також придатні для обґрунтування клінічних досліджень у Японії та США: Якщо імуногенність чи непереносимість перешкоджає проведенню довгострокових досліджень; При короткостроковій експозиції з багаторазовим введенням, навіть якщо тривалість клінічного дослідження перевищує 6 місяців, наприклад, при нерегулярному застосуванні при мігрені, еректильній дисфункції або простому герпесі; Лікарські препарати, які застосовуються довгостроково для зниження ризику рецидиву онкологічних захворювань; Лікарські препарати, які застосовуються за показаннями, для яких встановлена коротка очікувана тривалість життя. |
||
6.2 Державна реєстрація
Враховуючи велику кількість пацієнтів групи ризику та відносно менш контрольовані умови застосування препаратів у медичній практиці на відміну від клінічних досліджень, для обґрунтування можливості медичного застосування препарату необхідні доклінічні дослідження з більшою тривалістю, ніж для обґрунтування клінічних досліджень. Тривалість досліджень токсичності з багаторазовим введенням, необхідна для обґрунтування дозволу на медичне застосування препаратів з різною тривалістю лікування, наведена в таблиці 2. В окремих випадках для невеликої кількості патологічних станів, коли рекомендована тривалість застосування препарату становить від 2 тижнів до 3 місяців, але існує велика клінічний досвід, що передбачає більш широке та тривале клінічне застосування (наприклад, при тривожних станах, сезонному алергічному риніті, болі), можуть знадобитися токсикологічні дослідження з тривалістю, більшою за випадки, коли рекомендована тривалість застосування лікарського препарату перевищує 3 місяці.
Таблиця 2 – Рекомендована тривалість токсикологічних досліджень із багаторазовим введенням, необхідна для державної реєстрації лікарського засобу*
Тривалість застосування за показанням | Негризуни |
|
До двох тижнів | ||
Понад два тижні до одного місяця | ||
Понад один місяць до трьох місяців | 6 місяців | 6 місяців |
Понад три місяці | 6 місяців | 9 місяців |
* Роз'яснення наведено у примітках до таблиці 1. |
||
Визначення величини дози, що вперше вводиться людині, - важливий елемент забезпечення безпеки суб'єктів, що беруть участь у перших клінічних дослідженнях. При визначенні стартової дози, що рекомендується, для людини повинні бути оцінені всі значущі доклінічні дані, включаючи фармакологічні дозозалежні ефекти, фармакологічний/токсикологічний профіль, фармакокінетичні дані.
У цілому нині найважливішу інформацію дає висока нетоксична доза (ВНТД, NOAEL), встановлена у доклінічних дослідженнях безпеки найбільш відповідних видах тварин. Передбачувана клінічна стартова доза може залежати від різних факторів, включаючи фармакодинамічні параметри, індивідуальні властивості діючої речовини, а також дизайн клінічних досліджень. Окремі підходи представлені у національних керівництвах.
Пошукові клінічні дослідження (розділ 8) у людини можуть бути розпочаті при меншому або іншому обсязі доклінічних досліджень, ніж потрібно для досліджень клінічної розробки (6.1), у зв'язку з чим визначення клінічної стартової (та максимальної) дози може відрізнятися. Рекомендовані критерії вибору стартових доз у різних пошукових дослідженнях наведені в таблиці 3.
У ряді випадків наявність ранніх даних, отриманих у людини, може забезпечити краще розуміння фізіологічних/фармакологічних характеристик препарату для людини, властивостей лікарського препарату, що розробляється, і відповідності терапевтичних мішеней даному захворюванню. Раціональні ранні пошукові дослідження можуть вирішити такі завдання. У рамках цього стандарту під пошуковими клінічними дослідженнями розуміються дослідження, які проводяться на початку I фази, що передбачають обмежену експозицію і не передбачають оцінку терапевтичної ефективності та клінічної переносимості. Їх проводять для вивчення різних параметрів, таких як ФД, ФК препарату та інші біомаркери, які можуть включати рецепторне зв'язування та заміщення, що визначається за допомогою ПЕТ, або інші діагностичні параметри. Суб'єктами цих досліджень можуть бути як пацієнти з цільової популяції, і здорові добровольці.
У цих випадках обсяг та вид необхідних доклінічних даних залежатимуть від величини експозиції у людини з урахуванням максимальної клінічної дози та тривалості застосування. П'ять різних прикладів пошукових клінічних досліджень згруповані і детальніше описані далі і в таблиці 3, включаючи програми доклінічних досліджень, які можуть бути рекомендовані в цих випадках. Дозволяється використовувати альтернативні підходи, не описані в цьому стандарті, включаючи підходи для обґрунтування клінічних досліджень біотехнологічних лікарських препаратів. Альтернативні підходи до пошукових клінічних досліджень рекомендується обговорити та узгодити з відповідними регуляторними органами. Застосування будь-якого з цих підходів може призвести до зменшення використання лабораторних тварин при розробці лікарських засобів.
Рекомендовані вихідні дози та максимальні дози для використання в токсикологічних дослідженнях наведені в таблиці 3. У всіх випадках встановлення ФД та фармакологічних параметрів з використанням моделей in vivoта/або in vitroдуже важливо, як зазначено в таблиці 3 та розділі 2, і ці дані повинні використовуватися при обґрунтуванні вибраної дози для людини.
8.1 Клінічні дослідження з використанням мікродози
Наведені у цьому розділі два різні підходи з використанням мікродоз більш детально описані у таблиці 3.
При першому підході загальна доза препарату повинна бути не більше 100 мкг, які вводяться кожному суб'єкту дослідження одномоментно (одна доза) або кілька прийомів. Дослідження проводять з метою вивчення зв'язування рецепторів-мішеней або розподілу речовини у тканинах за допомогою ПЕТ. Також метою такого дослідження може бути вивчення ФК із використанням або без використання радіоактивної мітки.
При другому підході суб'єктам дослідження вводиться 5 і менше доз у кількості не більше 100 мг (загалом 500 мкг одному суб'єкту). Такі дослідження проводяться з аналогічними цілями, як і при використанні вищезгаданого підходу, але за наявності менш активних ПЕТ-лігандів.
У деяких випадках може бути прийнятним проведення клінічного дослідження з використанням мікродоз та внутрішньовенним введенням препарату, призначеного для прийому внутрішньо, та наявністю повного обсягу доклінічних токсикологічних даних для перорального шляху введення. При цьому внутрішньовенно мікродоза, що вводиться, може розглядатися за наявністю токсикологічних даних для перорального шляху введення, як описано в таблицях 1 і 3, як підхід 3, при якому були досягнуті прийнятні рівні експозиції. У цьому випадку не рекомендується досліджувати внутрішньовенну місцеву переносимість діючої речовини, оскільки доза, що вводиться, вкрай низька (не більше 100 мкг). Якщо у складі препарату, що вводиться внутрішньовенно, використовується новий розчинник, місцева переносимість розчинника повинна бути вивчена.
8.2 Клінічні дослідження з одноразовим введенням дози в діапазоні субтерапевтичних значень або в очікуваному терапевтичному діапазоні
При цьому підході (підхід 3) проводять клінічне дослідження з одноразовим введенням дози препарату, зазвичай починаючи з субтерапевтичних доз та згодом підвищуючи її до фармакологічно ефективного або очікуваного терапевтичного діапазону (див. таблицю 3). Визначення допустимої максимальної дози має бути засноване на доклінічних даних, але надалі вона може бути обмежена на основі отриманих в ході дослідження клінічних даних. Використання цього підходу може дозволити, наприклад, визначити параметри ФК із введенням препарату без радіоактивної мітки у прогнозованій фармакодинамічно ефективній дозі або близькій до неї. Іншим прикладом застосування цього підходу є оцінка на мета або фармакологічного впливу після одноразового введення. Дослідження із застосуванням цього підходу не призначені для обґрунтування максимальної клінічної дози, що переноситься (див. винятки, примітка "а" до таблиці 1).
8.3 Клінічні дослідження з використанням кількох доз
Для обґрунтування клінічних досліджень з використанням кількох доз для проведення доклінічних досліджень використовують два різні підходи (підходи 4 та 5 у таблиці 3). Дослідження на їх основі дозволяють обґрунтувати тривалість введення препаратів у дозах терапевтичного діапазону протягом 14 днів для оцінки параметрів ФК та ФД у людини, але їх не використовують для обґрунтування визначення максимальної клінічної дози, що переноситься.
При застосуванні підходу 4 проводиться двотижневе токсикологічне дослідження з багаторазовим введенням препарату гризунам та негризунам. Вибір дози, що вводиться тваринам, заснований на кратній дозі експозиції на рівні очікуваної AUC при максимальній клінічній дозі.
При використанні підходу 5 проводиться двотижневе токсикологічне дослідження на гризунах та підтверджує токсикологічне дослідження у негризунів, метою якого є підтвердження відсутності токсичного ефекту ВНТД (NOAEL) для гризунів при введенні негризунам. Якщо при введенні негризунам ВНТД (NOAEL) для гризунів спостерігається токсичний ефект, клінічне застосування препарату має бути відкладено до отримання даних подальших доклінічних досліджень у тварин цього виду (зазвичай стандартного токсикологічного дослідження, розділ 5).
Таблиця 3 - Доклінічні дослідження, що рекомендуються, для обґрунтування можливості проведення пошукових клінічних досліджень
Клінічні дослідження | Доклінічні дослідження |
|||
Дози, що вводяться | Вихідна та максимальна дози | Фармакологія | Загальнотоксичні дослідження | Дослідження генотоксич- |
Загальна доза 100 мкг (без інтервалу між прийомом дози), і загальна доза 1/100-а NOAEL і 1/100-а фармакологія | Вихідна та максимальна дози можуть бути однаковими, але не повинні перевищувати загальну дозу 100 мкг. | Профіль мішеней/рецепторів in vitroмає бути оцінений | Розширене токсикологічне дослідження з одноразовим введенням дози (див. примітки з і d) тваринам одного виду, як правило, гризуни, з пропонованим для використання в клініці шляху введення з отриманням токсикокінетичес- | Для ефективних радіоактивних міток (наприклад, міток для ПЕТ) повинні бути представлені відповідні |
Загальна кумулятивна доза 500 мкг, не більше 5 введень препарату з періодом відмивання між введеннями (6 і більше фактичних або прогнозу- | Вихідна та максимальна дози можуть бути однаковими, але не повинні перевищувати 100 мкг | Профіль мішеней/рецепторів in vitroмає бути оцінений Для обґрунтування вибору дози для застосування у людини повинні бути отримані з використанням фармакологічно релевантної моделі детальні дані про основні (первинні) фармакологічні параметри (механізм дії та/або ефекти) | Токсикологічне дослідження тривалістю 7 днів з багаторазовим введенням тваринам одного виду, як правило, гризунам, з пропонованим для використання в клініці шляху введення з отриманням токсикокінетичес- Повинні бути отримані гематологічні, лабораторні клінічні дані, дані некропії та гістопатологічного дослідження Може бути використана максимальна доза, 1000-кратна клінічна доза з перерахуванням в мг/кг для внутрішньовенного введення та мг/м для перорального шляху введення. | Проведення дослідження генотоксичності необов'язкове, але будь-які дослідження або оцінки SAR повинні бути включені до документів на отримання дозволу на проведення клінічного дослідження. Для ефективних радіоактивних міток (наприклад, міток для ПЕТ) мають бути представлені відповідні оцінки ФК параметрів міток та дозиметричні дані |
Дослідження з однією дозою в субтерапії | Вибір вихідної стартової дози повинен бути заснований на типах токсикологічних даних, отриманих у найбільш чутливих видів лабораторних тварин та даних про фармакологічно ефективну дозу. Також мають бути враховані національні рекомендації щодо вибору вихідної початкової дози для людини Максимальна доза може бути встановлена на рівні до 1/2 NOAEL експозиції у найбільш чутливих видів лабораторних тварин у випадках, коли прояв будь-якого значимого токсичного ефекту, зазначеного у тварин, можливий і оборотний у людини | Профіль мішеней/рецепторів in vitroмає бути оцінений Для обґрунтування вибору дози для застосування у людини повинні бути отримані з використанням фармакологічно релевантної моделі детальні дані про основні (первинні) фармакологічні параметри (механізм дії та/або ефекти) Основний набір фармакологічних досліджень безпеки (див. розділ 2) | Розширене токсикологічне дослідження з одноразовим введенням дози (див. примітки с) за можливим клінічним шляхом введення препарату з отриманням токсикокінетичних, гематологічних, лабораторних клінічних даних, даних некропсії та гістопатологічного дослідження. У цьому випадку як високу дозу використовують MTD, MFD або обмежено високу дозу (див. 1.1) |
|
Введення препарату протягом 14 днів у терапевти- | При прояві токсичних ефектів в обох видів лабораторних тварин слід виконувати національні вимоги щодо вибору початкової клінічної дози. Якщо токсичні ефекти не спостерігалися в жодного з видів лабораторних тварин (тобто NOAEL представляють найвищі досліджені дози в доклінічних дослідженнях, і використані дози не були нічим обмежені, наприклад не являють собою MFD) або відзначені тільки в одного виду лабораторних тварин, то стартової клінічної дозою повинна бути одна з доз, що забезпечує досягнення прогнозованого значення клінічної AUC (заснованого або на моделюванні ФК у різних видів, або на перерахунку мг/м), яка становить 1/50 AUC при використанні NOAEL у тварин і при якій отримували меншу експозицію За відсутності токсичних ефектів у обох видів тварин рекомендується використовувати максимальну клінічну дозу, що не перевищує 1/10 нижчої експозиції (AUC) у будь-якого виду, отриману у тварин будь-якого виду при введенні найвищої дози. Якщо токсичні ефекти спостерігаються тільки у одного виду тварин, максимальна клінічна доза не повинна перевищувати NOAEL для тварин того виду, у яких спостерігалися токсичні ефекти, або становити 1/2 AUC при введенні найвищої дози, що вводиться, при якій токсичні ефекти були відсутні (вибирається найменша із зазначених). ). За наявності токсичних ефектів в обох видів тварин вибір максимальної клінічної дози повинен бути заснований на стандартному підході оцінки ризиків, і в такому випадку може бути оцінена клінічна MTD. | Профіль мішеней/рецепторів in vitroмає бути оцінений Для обґрунтування вибору дози для застосування у людини повинні бути отримані з використанням фармакологічно релевантної моделі детальні дані про основні (первинні) фармакологічні параметри (механізм дії та/або ефекти) Основний набір фармакологічних досліджень безпеки (див. розділ 2) з використанням доз, аналогічних дозам загальнотоксико- | Токсикологічне дослідження тривалістю 14 днів з багаторазовим введенням гризунам та негризунам зі стандартним набором оцінюваних параметрів; вибір дози, що використовується, заснований на кратності експозиції очікуваної клінічної AUC при максимальній дозі. | Тест Еймса (або альтернативний тест, якщо проведення тесту Еймса не прийнятно, наприклад для антибакте- |
Введення препарату протягом 14 днів, без перевищення продовження | Прогнозована експозиція при введенні стартових доз не повинна перевищувати 1/50 NOAEL у найбільш чутливих видів тварин у розрахунку мг/м. Слід враховувати національні рекомендації щодо вибору початкової клінічної дози Максимальна експозиція у людини не повинна перевищувати AUC при NOAEL у негризунів або 1/2 AUC при NOAEL у гризунів (вибирається найменша із зазначених) | Профіль мішеней/рецепторів in vitroмає бути оцінений Для обґрунтування вибору дози для застосування у людини повинні бути отримані з використанням фармакологічно релевантної моделі детальні дані про основні (первинні) фармакологічні параметри (механізм дії та/або ефекти) Основний набір фармакологічних досліджень безпеки (див. розділ 2) з використанням доз, аналогічних дозам у загальнотоксикологічних дослідженнях | Стандартне токсикологічне дослідження тривалістю 14 днів із багаторазовим введенням гризунам (з обґрунтуванням вибору гризунів як прийнятного виду лабораторних тварин для даного дослідження). Як високу дозу використовують MTD, MFD або обмежено високу дозу (див. 1.1) Підтверджує дослідження у негризунів n=3) в очікуваній експозиції NOAEL для гризунів тривалістю не менше 3 днів і найменшою тривалістю передбачуваного клінічного дослідження Може бути проведено альтернативне дослідження з ескалацією дози у негризунів тривалістю не менше 3 днів та найменшою тривалістю передбачуваного клінічного дослідження при введенні дози для досягнення NOAEL експозиції для гризунів. | Тест Еймса (або альтернативний тест, якщо проведення тесту Еймса не прийнятно, наприклад для антибактері- |
Загальнотоксичні доклінічні дослідження повинні проводитись відповідно до правил GLP. План дослідження генотоксичності та вибір дози описані в посібнику ICH S2B. План розширеного дослідження з одноразовим введенням дози, як правило, повинен включати оцінку гематологічних, лабораторних клінічних даних, даних некропсії та гістопатологічного дослідження (вводяться лише контрольна та висока дози, якщо токсичні ефекти препарату не спостерігаються при введенні високої дози) після одноразового введення, з наступним спостереженням протягом двох тижнів для оцінки відстрочених токсичних ефектів та їх зникнення. Стандартний дизайн дослідження на гризунах включає проведення токсикологічної оцінки на 10 тварин/стат/група через день після введення препарату, на 5 тварин/стат, що отримували обрані дози (дозу), що оцінюються на 14-й день після введення. Стандартний дизайн дослідження на негризунах включає проведення оцінки на 3 тварини/підлогу/група для всіх груп на 2-й день після введення препарату і на 2 тварин/статі, що отримували обрані дози (дозу), оцінювані на 14-й день після введення. Рівень однієї дози для оцінки оборотності/відстрочення токсичних ефектів на 14 день після введення може використовуватися для обґрунтування підходу з використанням мікродоз. Використовуваний рівень дози, що вводиться тваринам, не слід встановлювати на рівні високої дози, це має бути доза щонайменше 100-кратної клінічної дози. За відсутності розвитку небажаних ефектів при проведенні клінічних досліджень ескалація доз вище за цю AUC може бути прийнятною, якщо дані токсикологічних досліджень дозволяють вважати можливі небажані ефекти у людини, що виявляються, оборотними та низькою тяжкістю. |
||||
Місцеву переносимість при запропонованому способі введення у клінічних дослідженнях переважно вивчати у рамках дослідження загальнотоксичної дії; окремі дослідження зазвичай проводити не рекомендується.
Для обґрунтування обмежених клінічних досліджень альтернативного за терапевтичним способом введення (наприклад, одноразове внутрішньовенне введення з метою визначення абсолютної біодоступності лікарського препарату, що приймається внутрішньо) допустимо вивчення переносимості одноразової дози на одному виді тварин. У випадках, коли очікувана системна експозиція (AUC та С) для нетерапевтичного способу введення вивчена в рамках вже проведених токсикологічних досліджень, кінцеві точки дослідження місцевої переносимості можуть бути обмежені клінічними ефектами та макро- та мікроскопічним обстеженням місця введення. Склад лікарського препарату, призначеного для вивчення місцевої переносимості, може не бути ідентичним, але повинен бути подібним до складу та лікарської форми лікарського препарату, що використовується в клінічних дослідженнях.
Для внутрішньовенного мікродозового дослідження, яке проводиться за наявності токсикологічних даних для перорального введення (див. розділ 7), оцінка місцевої переносимості фармацевтичної субстанції не потрібна. Якщо у складі препарату для внутрішньовенного введення використовуватиметься новий розчинник, необхідно вивчити його місцеву переносимість.
Для парентеральних лікарських засобів вивчення місцевої переносимості у непередбачених місцях ін'єкцій, якщо потрібно, необхідно провести до призначення лікарського препарату велику кількість пацієнтів (наприклад, до III фази клінічних досліджень). Підхід до планування таких досліджень у різних країнах різний. Такі дослідження не потрібні в США (прикладом виключення може бути інтратекальне введення при запланованому епідуральному введенні). У Японії та країнах ЄС рекомендується одноразове паравенозне введення для внутрішньовенного способу. Необхідність дослідження інших парентеральних шляхів запровадження оцінюється індивідуально.
Для обґрунтування всіх клінічних досліджень із одноразовим введенням лікарського препарату достатнім вважається проведення тесту на генні мутації. Для обґрунтування клінічних досліджень із багаторазовим введенням лікарського препарату необхідні додаткові дослідження, що дозволяють виявити хромосомні ушкодження у ссавців. Повний набір тестів і генотоксичність необхідно виконати до початку II фази клінічних досліджень.
Якщо результати дослідження свідчать про наявність генотоксичної дії, необхідно їх оцінити і, можливо, провести додаткові дослідження для встановлення прийнятності подальшого застосування лікарського препарату для людини.
Дослідження генотоксичності, що рекомендуються для обґрунтування пошукових клінічних досліджень з використанням різних підходів, обговорюються у розділі 8 цього стандарту.
Випадки, що вимагають вивчення канцерогенності, обговорюються в посібнику ICH S1A щодо оцінки необхідності вивчення канцерогенності лікарських препаратів. У зазначених випадках дослідження канцерогенності мають бути проведені на початок процедури державної реєстрації. У випадках, коли є вагомі причини, що вказують на канцерогенний ризик, результати досліджень необхідно подати перед проведенням клінічних досліджень. Велика тривалість клінічного дослідження не розглядається як обов'язкова причина для досліджень канцерогенності.
Необхідні дослідження канцерогенності лікарських засобів, розроблюваних для лікування серйозних захворювань у дорослих і дітей, допускається за погодженням з регуляторним органом проводити після їх державної реєстрації.
Дослідження репродуктивної токсичності необхідно проводити, беручи до уваги популяцію пацієнтів, яка застосовуватиме досліджуваний лікарський препарат.
12.1 Чоловіки
Чоловіки можуть бути включені до клінічних досліджень фази I та фази II до проведення оцінки репродуктивної системи самців через те, що оцінка репродуктивної системи самців проводиться в дослідженнях токсичності при багаторазовому введенні.
Примітка 2 - Оцінка фертильності самців і самок за стандартним гістологічним дослідженням яєчок і яєчників у дослідженнях токсичності (при багаторазовому введенні зазвичай гризунам) не менше 2-тижневої тривалості за здатністю виявляти токсичні ефекти вважається порівнянною з дослідженнями фертильності для виявлення самок.
Дослідження фертильності у самців мають бути завершені до початку великомасштабних чи тривалих клінічних досліджень (наприклад, досліджень ІІІ фази).
12.2 Жінки, які не мають дітородного потенціалу
Якщо проведені відповідні дослідження токсичності при багаторазовому введенні (які включають оцінку репродуктивних органів самок), допускається включати до клінічних досліджень жінок, які не мають дітородного потенціалу (тобто перманентно стерилізовані, які перебувають у постменопаузі), без досліджень репродуктивної токсичності. Постменопауза визначається відсутність менструацій протягом 12 місяців без інших медичних причин.
12.3 Жінки, які мають дітородний потенціал
Для жінок, які мають дітородний потенціал (ЖСДП, WOCBP), існує високий ризик ненавмисного впливу лікарського препарату на ембріон або плід до отримання інформації про співвідношення потенційної користі та ризику. У всіх країнах, що застосовують керівництво ICH, існують подібні рекомендації щодо термінів проведення досліджень репродуктивної токсичності для включення ЖСДП до клінічних досліджень.
При включенні до дослідження ЖСДП необхідно визначити та мінімізувати ризик ненавмисного впливу на ембріон або плід. Перший підхід до досягнення поставленої мети полягає у проведенні досліджень репродуктивної токсичності для оцінки ризику застосування лікарського препарату та вживання належних запобіжних заходів у клінічних дослідженнях у ЖСДП. Другий підхід полягає в обмеженні ризиків шляхом вжиття заходів щодо запобігання настанню вагітності під час клінічних досліджень. До таких заходів належать тести на вагітність (наприклад, з вільної (3-субодиниці ХГЛ), використання високонадійних методів контрацепції (примітка 3) та включення до дослідження тільки після підтвердження наявності менструацій. Тести на вагітність, що проводяться під час клінічного дослідження, та навчання пацієнтів повинні бути достатніми, щоб забезпечити виконання заходів, спрямованих на попередження вагітності в період експозиції лікарського препарату (який може перевищувати тривалість дослідження). препаратів, що мають подібну будову, або фармакологічні ефекти Якщо значуща інформація про вплив на репродукцію відсутня, необхідно інформувати пацієнта про потенційний невиявлений ризик для ембріона чи плода.
У всіх країнах, що застосовують керівництво ICH, за певних умов допускається включати ЖСДП до клінічних досліджень ранніх фаз без доклінічного вивчення онтогенетичної токсичності (наприклад, без дослідження можливого впливу на ембріональний та фетальний розвиток). Однією з таких умов є належний контроль ризику настання вагітності під час короткострокових (наприклад, 2-тижневих) клінічних досліджень. Іншою умовою може бути переважання захворювання серед жінок, коли неможливо досягти мети дослідження без включення ЖСДП, при цьому вжито достатніх заходів щодо запобігання вагітності (див. вище).
Примітка 3 - Високонадійними методами контрацепції вважаються як поодинокі, так і комбіновані препарати, що забезпечують низьку частоту настання вагітності (тобто менше 1% на рік) при їх постійному та правильному застосуванні. Для пацієнтів, які застосовують гормональні контрацептивні препарати, необхідно надати інформацію про вплив досліджуваного лікарського препарату на контрацепцію.
Додатковим обґрунтуванням можливості проведення досліджень у ЖСДП без доклінічних досліджень онтогенетичної токсичності є відомості про механізм дії лікарського препарату, його властивості, тривалість впливу на плід або складність проведення досліджень онтогенетичної токсичності на відповідній моделі тварин. Наприклад, для моноклональних антитіл, які, за наявними сучасними науковими даними, мають слабкий ембріональний та фетальний вплив у період органогенезу, дослідження онтогенетичної токсичності допускається проводити під час III фази клінічних досліджень. Звіт про завершене дослідження необхідно подати у складі реєстраційного досьє.
Загалом за наявності попередніх даних про репродуктивну токсичність на двох видах тварин (примітка 4) та при вжитті заходів щодо попередження вагітності (див. вище) допускається включення ЖСДП (до 150 суб'єктів), які отримують досліджуваний лікарський препарат протягом відносно короткого періоду (до 3 місяців) до проведення спеціальних досліджень репродуктивної токсичності. Підставою для цього є дуже низька частота настання вагітності у контрольованих дослідженнях такого розміру та тривалості (примітка 5) та здатність належним чином спланованих попередніх досліджень виявити найважливіші онтогенетичні токсичні ефекти, які можуть виявити ризики при включенні ЖСДП до клінічних досліджень. На кількість ЖСДП, що включаються до дослідження, та тривалість дослідження можуть вплинути характеристики популяції, які знижують ймовірність настання вагітності (наприклад, вік, захворювання).
Примітка 4 - Якщо дози адекватні, для досягнення цієї мети доцільно попереднє дослідження ембріонального та фетального розвитку, яке включає оцінку виживання плодів, маси тіла, зовнішнє вивчення та вивчення внутрішніх органів з використанням не менше шести самок на групу за наявності самок, які отримували лікарський препарат період органогенезу. Такі попередні доклінічні дослідження повинні проводитися за високими науковими стандартами з простим доступом до даних або відповідно до вимог GLP.
Примітка 5 - Частота настання вагітності у жінок, які вперше роблять спробу завагітніти, становить приблизно 17% на менструальний цикл. Частота настання вагітності в дослідженнях III фази, проведених у жінок, які мають дітородний потенціал, становить<0,1% на менструальный цикл. В ходе этих исследований пациентов следует предупредить о нежелательности наступления беременности и необходимости соблюдения мер по предупреждению беременности. По имеющимся данным, частота наступления беременности во II фазе ниже, чем в III фазе, но в силу ограниченного количества включенных женщин величину снижения установить невозможно. Основываясь на данных III фазы, частота наступления беременности во II фазе исследований, включающих 150 женщин с сохраненным детородным потенциалом и продолжительностью до 3 месяцев, значительно меньше 0,5 беременностей на лекарственный препарат, находящийся в разработке.
У США дослідження ембріонального та фетального розвитку можуть бути відстрочені до III фази досліджень із включенням ЖСДП при вжитті заходів щодо попередження вагітності (див. вище). У ЄС та Японії (за винятком ситуацій, описаних вище в цьому розділі), спеціальні дослідження онтогенетичної токсичності необхідно завершити до включення до дослідження ЖСДП.
У всіх країнах, що застосовують керівництво ICH, допускається включати ЖСДП до клінічних досліджень І та ІІ фаз з багаторазовим введенням лікарського препарату до проведення дослідження фертильності у самок, враховуючи те, що оцінка органів репродуктивної системи у тварин здійснюється в рамках досліджень токсичності при багаторазовому введенні (примітка 2). Для включення ЖСДП у великомасштабні та тривалі клінічні дослідження (наприклад, дослідження фази III) необхідно провести спеціальні доклінічні дослідження фертильності у самок.
У всіх країнах, що застосовують керівництво ICH, для державної реєстрації лікарського засобу необхідно подати результати досліджень пре- та постнатального одногенетичного розвитку.
До включення до будь-яких досліджень ЖСДП, які не користуються високоефективними методами контрацепції (примітка 3) або з невідомим гестаційним статусом, потрібне подання даних завершеного дослідження репродуктивної токсичності та стандартний набір тестів на генотоксичність.
12.4 Вагітні жінки
До включення вагітних жінок до клінічних досліджень необхідно провести повне дослідження репродуктивної токсичності та стандартний набір тестів на генотоксичність. Крім цього, необхідно оцінити наявні дані щодо безпеки застосування лікарського препарату для людини.
При обґрунтуванні можливості включення до клінічних досліджень педіатричних пацієнтів найбільш значну інформацію надають дані щодо безпеки з попередніх досліджень на дорослих пацієнтах - вона має бути доступна до початку досліджень у дітей. Достатність та обсяг даних клінічних досліджень у дорослих для цього рішення визначаються в індивідуальному порядку. До початку застосування у дітей достатні дані про досвід застосування у дорослих можуть бути відсутніми (наприклад, при виключно педіатричних показаннях до застосування).
До початку досліджень у дітей мають бути отримані результати досліджень токсичності при багаторазовому введенні відповідної тривалості на дорослих тварин (див. таблицю 1), проведено основний набір досліджень фармакологічної безпеки та стандартний набір тестів на генотоксичність. Також можуть бути необхідні дані репродуктивної токсичності, що відповідають віку та статі досліджуваних дітей, що надають інформацію про прямий токсичний ризик або вплив на розвиток (наприклад, дослідження фертильності, пре- та постнатального розвитку). Дослідження ембріонального та фетального розвитку не є критичними для обґрунтування можливості проведення клінічних досліджень у пацієнтів чоловічої статі чи препубертатних пацієнтів жіночої статі.
Необхідність проведення будь-яких досліджень на нестатевозрілих тваринах слід розглядати, тільки якщо попередні дані у тварин та дані з безпеки у людини, включаючи ефекти інших лікарських препаратів даного фармакологічного класу, розцінюються як недостатні для обґрунтування можливості проведення клінічного дослідження у дітей. Якщо таке доклінічне дослідження необхідне, достатньо використання одного виду тварин, переважно гризунів. За достатнього наукового обґрунтування допускається дослідження на негризунах.
Для короткострокових ФК-досліджень у дітей (наприклад, 1-3 дози) дослідження токсичності на нестатевозрілих тварин, як правило, не вважаються інформативними.
Залежно від показань до застосування, віку дітей, що включаються до клінічного дослідження, та даних щодо безпеки застосування для дорослих тварин та пацієнтів слід розглянути питання про необхідність отримання результатів досліджень на нестатевих зрілих тварин до початку короткострокових клінічних досліджень ефективності з використанням великого діапазону доз та безпеки лікарського засобу. препарату. Однією з найважливіших питань є вік учасників дослідження стосовно тривалості дослідження (тобто частка періоду розвитку, протягом якого учасники дослідження приймають лікарський препарат). Даний фактор є визначальним в оцінці необхідності проведення доклінічних досліджень на нестатевозрілих тварин, і, якщо вони необхідні, слід встановити терміни їх проведення стосовно клінічних досліджень.
До початку тривалих клінічних досліджень у педіатричних пацієнтів, для обґрунтування яких знадобилося проведення дослідження токсичності на нестатевозрілих тварин, ці доклінічні дослідження мають бути завершені.
Можливі ситуації, коли педіатричні пацієнти є основною терапевтичною популяцією, а наявні експериментальні дані вказують на потенційний вплив досліджуваного препарату на розвиток органів-мішеней (фармакологічний токсикологічний плі). У деяких із цих випадків можуть знадобитися тривалі дослідження на нестатевозрілих тварин. Допустимим є тривале токсикологічне дослідження на тваринах відповідного виду та віку (наприклад, 12-місячне дослідження на собаках або 6-місячне дослідження на щурах). 12-місячне дослідження може охоплювати весь період розвитку собак. Для інших видів лабораторних тварин цей дизайн можна адаптувати для заміни відповідного стандартного хронічного дослідження та окремого дослідження на нестатевозрілих тварин за певних умов.
На початок тривалих клінічних досліджень в дітей віком потрібно визначити необхідність вивчення канцерогенності. Однак, якщо суттєвих підстав (наприклад, доказів гепатотоксичності з різних тестів або наявності проканцерогенного ризику, зумовленого механізмом дії або ефектами, виявленими при вивченні загальнотоксичної дії) відсутні, вивчення канцерогенності для проведення клінічних досліджень у дітей не потрібно.
Як зазначено в посібнику ICH S8 з досліджень імунотоксичності лікарських препаратів, всі нові лікарські препарати підлягають оцінці на наявність імунотоксичного потенціалу з використанням стандартних токсикологічних досліджень і додаткових досліджень імунотоксичності, що проводяться на підставі огляду сукупності доказів, включаючи імуноопосередковані сигнали. Якщо існує необхідність проведення додаткових досліджень імунотоксичності, їх необхідно завершити до початку застосування лікарського препарату, що досліджується, у великих популяцій пацієнтів (наприклад, клінічні дослідження III фази).
Необхідність чи час проведення дослідження фотобезпеки залежно від експозиції у людини визначаються:
- фотохімічними властивостями (наприклад, фотоабсорбцією та фотостабільністю) молекули;
- інформацією про фототоксичний потенціал хімічно подібних сполук;
- розподілом у тканинах;
- клінічними чи доклінічними даними, що вказують на наявність фототоксичності.
Повинна бути проведена початкова оцінка фототоксичного потенціалу, що ґрунтується на фотохімічних властивостях лікарського препарату та його фармакологічному/хімічному класі. Якщо оцінка всіх доступних даних та передбачуваного плану клінічних досліджень вказує на значний ризик фототоксичності для людини, необхідно передбачити заходи захисту пацієнта під час амбулаторних клінічних досліджень. Крім того, для надання відомостей про ризик для людини та потреби у подальшому вивченні необхідно виконати наступну доклінічну оцінку розподілу діючої речовини у шкірі та очах. Тоді, якщо можна застосувати, експериментальну оцінку (доклінічна, in vitroабо in vivo, або клінічна) фототоксичного потенціалу слід провести до початку застосування лікарського препарату до великої кількості пацієнтів (клінічні дослідження ІІІ фази).
Альтернативно, замість вищеописаного покрокового підходу можна провести пряму оцінку фототоксичного потенціалу в доклінічних або клінічних дослідженнях. Якщо результати цих досліджень є негативними, то рання оцінка розподілу лікарського препарату в очах/шкірі та профілактичні заходи при проведенні клінічного дослідження не потрібні.
Якщо результати оцінки фототоксичності вказують на можливий фотоканцерогенний потенціал, у пацієнтів цей ризик зазвичай належним чином контролюється за допомогою захисних заходів, що включають попередження в інформованій згоді та інструкції щодо застосування (див. примітку 6).
Примітка 6 - Вивчення фотоканцерогенності на негризунах з використанням доступних зараз моделей (наприклад, безволосих гризунів) при розробці лікарських препаратів вважається недоцільним і, як правило, не потрібно. Якщо дослідження фототоксичності вказують на можливий фотоканцерогенний ризик і стає доступним відповідний метод вивчення, дослідження зазвичай необхідно завершити до початку процедури державної реєстрації, а його результати необхідно враховувати в оцінці ризику для людини.
Для лікарських засобів, що впливають на ЦНС, незалежно від показань до застосування, потрібно визначити необхідність оцінки ризику розвитку лікарської залежності. Доклінічні дослідження необхідні для обґрунтування дизайну клінічних досліджень, визначення особливої категорії, що застосовується в країні (наприклад, списки наркотичних та психотропних речовин та ін.), та складання інструкції щодо застосування. При формуванні набору необхідних досліджень слід керуватися національними посібниками з доклінічної оцінки, ризик розвитку лікарської залежності.
Доклінічні дані, зібрані на ранніх етапах розробки лікарського засобу, можуть бути інформативними щодо виявлення ранніх індикаторів потенціалу розвитку залежності. Дані про такі ранні індикатори повинні бути отримані до першого застосування лікарського засобу до людини; вони включають ФК/ФД-профіль для визначення тривалості дії, подібності хімічної структури з лікарськими препаратами, що викликають розвиток лікарської залежності, профіль рецепторного зв'язування та поведінкові/клінічні симптоми з доклінічних досліджень in vivo.Якщо за результатами цих ранніх досліджень потенціалу розвитку лікарської залежності не виявлено, розширене доклінічне вивчення на моделях лікарської залежності може не знадобитися. Як правило, якщо діюча речовина виявляє ознаки, подібні до відомих патернів лікарської залежності, або у нього є новий механізм дії на ЦНС, перед початком великих клінічних досліджень (наприклад, клінічні дослідження III фази) рекомендується проведення подальших доклінічних досліджень.
Якщо профіль метаболітів і мета впливу лікарського препарату у гризунів відповідає таким у людини, доклінічну оцінку ризику розвитку лікарської залежності проводять на гризунах. Нелюдоподібних приматів допускається використовувати тільки в тих поодиноких випадках, коли є незаперечні докази, що такі дослідження дозволять прогнозувати схильність людини до лікарської залежності, а моделі на гризунах неадекватні. Для оцінки ризику розвитку лікарської залежності найчастіше проводяться три види досліджень: перевага лікарського препарату, самостійне введення лікарського препарату та оцінка стану після його відміни. Дослідження переваги та самостійного введення лікарського препарату зазвичай проводяться як окремі експерименти. Дослідження з оцінкою скасування іноді можуть бути включені до дослідження токсичності при багаторазовому введенні (група оцінки оборотності токсичної дії). Максимальна доза, яка дозволяє досягти концентрації в плазмі крові лабораторних тварин, що у кілька разів перевищує таку для терапевтичної клінічної дози у людини, розглядається як відповідна для таких доклінічних оцінок ризику розвитку лікарської залежності.
Якщо раніше отримані доклінічні або клінічні дані про лікарський препарат або подібні лікарські препарати вказують на ймовірність особливих проблем щодо безпеки, можуть бути потрібні додаткові доклінічні дослідження (наприклад, для визначення потенційних біомаркерів, для з'ясування механізму дії).
У рекомендаціях ICH Q3A та Q3B представлені підходи до кваліфікації домішок та продуктів розкладання діючої речовини. Якщо для кваліфікації домішок і продуктів розпаду необхідні спеціальні дослідження, їх проведення, як правило, не потрібно до початку клінічних досліджень III фази, за винятком випадків, при яких зміни, що вносяться в ході розробки, призводять до появи практично нового профілю домішок (наприклад, нові шляхи синтезу , нові продукти розкладання, що утворюються внаслідок взаємодії між компонентами лікарського засобу). У таких випадках для обґрунтування проведення клінічних досліджень ІІ фази або пізніших етапів розробки можуть знадобитися відповідні дослідження з кваліфікації домішок та продуктів розпаду.
Цей розділ поширюється на комбіновані лікарські препарати, які призначені для одночасного застосування та вкладаються в одну упаковку або для прийому в одній лікарській формі ("фіксована комбінація"). Наведені нижче принципи також можуть бути застосовані до некомбінованих лікарських препаратів, які згідно з інструкцією із застосування можуть застосовуватися одночасно з певним лікарським препаратом, у тому числі і не у вигляді "фіксованої комбінації", а також для лікарських препаратів, які не мають достатніх клінічних даних щодо комбінованого застосування.
Цей стандарт застосовують до наступних комбінацій:
1) дві або більше речовин, що знаходяться на пізніх етапах розробки (сполуки зі значним досвідом клінічного застосування (тобто III фаза клінічного дослідження або післяреєстраційні дослідження);
2) одна або більше речовин, що знаходяться на пізніх етапах розробки, і одна або більше речовин, що знаходяться на ранніх етапах розробки (є обмежений досвід клінічного застосування, наприклад клінічне дослідження II фази і більш ранні фази дослідження), або
3) більше однієї речовини, що є на ранніх етапах розробки.
Для більшості комбінацій, що містять дві речовини, що знаходяться на пізніх етапах розробки, але для яких відсутній значний клінічний досвід спільного застосування, для обґрунтування можливості проведення клінічних досліджень або державної реєстрації не потрібне проведення комбінованих токсикологічних досліджень, якщо немає причин припускати можливий спільний токсикологічний ефект (наприклад наявність однакових для токсичного ефекту органів-мішеней). Зазначені причини можуть змінюватись в залежності від рівня безпеки та можливості моніторингу небажаних ефектів у людини. Якщо доклінічне дослідження потрібно для оцінки можливих спільних токсикологічних ефектів комбінації, воно має бути завершено до початку проведення клінічних досліджень комбінації.
Для комбінацій, що містять дві речовини, що знаходяться на пізніх етапах розробки, але для яких відсутній прийнятний клінічний досвід спільного застосування, для обґрунтування можливості проведення короткострокових клінічних досліджень (наприклад, дослідження II фази тривалістю до 3 місяців) доклінічні дослідження комбінації, як правило, не потрібні, якщо думка про відсутність можливих токсикологічних ефектів комбінації ґрунтується на достатніх наявних даних. Водночас для тривалих та великомасштабних клінічних досліджень, а також для процедури державної реєстрації проведення доклінічних досліджень подібних комбінацій є обов'язковим.
Для комбінацій речовин, що знаходяться на ранніх етапах розробки з клінічним досвідом застосування, з речовинами, що знаходяться на пізніх стадіях розробки, для комбінації яких відсутні суттєві токсикологічні побоювання, токсикологічні дослідження комбінації для обґрунтування можливості досліджень по "перевірці клінічної концепції" тривалістю до 1 місяця . Клінічні дослідження комбінації не повинні перевищувати за тривалістю клінічний досвід застосування окремих компонентів. Для клінічних досліджень пізніших стадій та більшої тривалості доклінічні дослідження комбінацій є обов'язковими.
Для комбінацій, що містять речовини, що знаходяться на ранніх етапах розробки, проведення доклінічних досліджень їх комбінації для обґрунтування можливості проведення клінічних досліджень необхідно.
Якщо для кожного з компонентів комбінації проведено повну програму доклінічних досліджень, а доклінічне токсикологічне вивчення комбінації необхідно для обґрунтування можливості проведення клінічного дослідження, тривалість дослідження комбінації має бути еквівалентною тривалості клінічного дослідження (але не більше 90 днів). Також це доклінічне дослідження буде придатним для процедури державної реєстрації. Доклінічне дослідження комбінації меншої тривалості також може бути прийнятним для процедури державної реєстрації залежно від тривалості передбачуваного клінічного застосування.
Дизайн доклінічних досліджень, що рекомендуються для вивчення комбінації, залежить від фармакологічного, токсикологічного та фармакокінетичного профілів окремих компонентів, показань до застосування, запропонованої цільової популяції пацієнтів та наявних клінічних даних.
Доклінічні дослідження комбінації зазвичай проводяться одному відповідному вигляді тварин. При виявленні несподіваних токсичних ефектів може знадобитися проведення додаткових досліджень.
У тих випадках, коли повна програма доклінічних досліджень для окремих компонентів не виконана, допускається проведення повної доклінічної токсикологічної програми лише щодо комбінації за умови, що окремі компоненти призначені лише для застосування у комбінації.
Якщо окремі компоненти були вивчені відповідно до чинних стандартів, то для проведення клінічних досліджень або процедури державної реєстрації дослідження генотоксичності, фармакологічної безпеки та канцерогенності комбінації, як правило, не потрібні. У випадках, коли популяція пацієнтів включає ЖСДП, а дослідження окремих компонентів (компонента) вказують на ембріональний та фетальний ризик, дослідження комбінації не рекомендуються, оскільки потенційна шкода для ембріонального та фетального розвитку людини вже була встановлена. Якщо доклінічні дослідження ембріонального та фетального розвитку вказують на те, що жоден з компонентів не несе ризику онтогенетичного розвитку людини, дослідження комбінації не потрібні за відсутності побоювань, що ґрунтуються на властивостях окремих компонентів, що їхня комбінація може мати ризики безпеки для людини. У випадках, коли вплив окремих компонентів композиції на ембріональний та фетальний розвиток вивчено, але потрібні дослідження комбінації, результати останніх необхідно подати до початку процедури державної реєстрації.
Скорочення
Area Under the Curve | Площа під фармакокінетичною кривою |
|
Maximum Plasma Concentration | Максимальна концентрація у плазмі крові |
|
Європейський Союз |
||
Good Laboratory Practices | Належна лабораторна практика |
|
Human Chorionic Gonadotropin | Хоріонічний гонадотропін людини |
|
Human Immunodeficiency Virus | Вірус імунодефіциту |
|
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use | Міжнародна конференція з гармонізації технічних вимог для реєстрації лікарських засобів для медичного застосування |
|
Внутрішньовенний |
||
Maximum Feasible Dose | Максимальна допустима доза |
|
Maximum Tolerated Dose | Максимальна доза, що переноситься |
|
ВНТД (NOAEL) | No Observed Adverse Effect Level | Висока нетоксична доза |
Positron Emission Tomography | Позитронно-емісійна томографія |
|
Pharmacokinetics | Фармакокінетика |
|
Pharmacodynamics | Фармакодинаміка |
|
Structure-Activity Relationship | Взаємозв'язки, зумовлені активністю молекулярної структури |
|
Small Interfering RNA | Мала інтерферуюча РНК |
|
ЖСДП (WOCBP) | Women of Childbearing Potential | Жінки, які мають дітородний потенціал |
ICH S6 Guideline: Preclinical Safety Evaluation for Biotechnological-Derived Pharmaceuticals; July 1997.
ICH E8 Guideline: General Considerations for Clinical Trials; July 1997.
ICH S5(R2) Guideline: Визначення токсичності для репродукції для медичних продуктів і токсичності для мінеральної продуктивності; June 1993 року.
ICH S1 C(R2) Guideline: Dose Selection for Carcinogenicity Studies of Pharmaceuticals; Березень 2008.
ICH S7A Guideline: Safety Pharmacology Studies for Human Pharmaceuticals; Листопад 2000.
ICH S7B Guideline: Nonclinical Evaluation of Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) By Human Pharmaceuticals; May 2005 року.
ICH S3A Guideline: Знайти про Guidance on Toxicokinetics: Зображення з Systemic Exposure in Toxicity Studies; Жовтень 1994 року.
Національний центр для відновлення, Refinement and Reducction of Animals in Research. Challenging Requirements for Acute Toxicity Studies: Workshop Report; May 2007 року.
Robinson S., Delongeas JL., Donald E., Dreher D., Festag M., Kervyn S. et al. A Європейський фармацевтичний фірма ініціативна розвідка regulatory requirement for acute toxicity studies в фармацевтичної drug development. Regul Toxicol Pharmacol 2008; 50: 345-352.
ICH S2B Guideline: Genotoxicity: A Standard Battery for Genotoxicity Testing for Pharmaceuticals; July 1997.
ICH S1A Guideline: Guideline на необхідності для кардиногенності Studies of Pharmaceuticals; November 1995.
ICH Q3A(R2) Guideline: Impurities in New Drug Substances; Жовтень 2006.
ICH Q3B(R2) Guideline: Impurities in New Drug Products; June 2006.
ICH S8 Guideline: Immunotoxicity Studies for Human Pharmaceuticals; September 2005.
Sakai Т., Takahashi M., Mitsumori K., Yasuhara K., Kawashima K., Mayahara H. et al. Collaborative work to evaluate toxicity on male reproductive organs 2-week repeated-dose toxicity studies in rats. Попередня сторінка. J Toxicol Sci 2000; 25:1-21.
Sanbuissho A., Yoshida M., Hisada S., Sagami F., Kudo S., Kumazawa T. et al. Collaborative work on evaluation of ovarian toxicity за допомогою реабілітаційної та fertility studies в female rats. J Toxicol Sci 2009; 34:1-22.
Додаток ТАК
(довідкове)
Таблиця ТАК.1
Позначення посилального міжнародного документа | Ступінь відповідності | Позначення та найменування відповідного національного стандарту |
ICH S3A Guideline | ГОСТ Р 56702-2015 "Лікарські засоби для медичного застосування. Доклінічні токсикологічні та фармакокінетичні дослідження безпеки" |
|
ICH S6 Guideline "Лікарські засоби для медичного застосування. Доклінічні фармакологічні дослідження безпеки" |
||
OECD Principles of good laboratory practice | ГОСТ Р 53434-2009 "Принципи належної лабораторної практики" |
|
Примітка - У цій таблиці використані такі умовні позначення ступеня відповідності стандартів: IDT – ідентичні стандарти; MOD – модифіковані стандарти. |
||
УДК 615.038:615.012/.014:615.2:006.354 | |
Ключові слова: лікарські засоби, доклінічні дослідження безпеки, клінічні дослідження, державна реєстрація, безпека |
|
Електронний текст документа
підготовлений АТ "Кодекс" і звірений за:
офіційне видання
М: Стандартінформ, 2016
Клінічне дослідження (КІ) -це вивчення клінічних, фармакологічних, фармакодинамічних властивостей досліджуваного препарату у людини, включаючи процеси всмоктування, розподілу, зміни та виведення, з метою отримання науковими методами оцінок та доказів ефективності та безпеки лікарських засобів, даних про очікувані побічні явища та ефекти взаємодії з іншими лікарськими засобами.
Метою КІ лікарських засобівє отримання науковими методами оцінок та доказів ефективності та безпеки лікарських засобів, даних про очікувані побічні ефекти від застосування лікарських засобів та ефекти взаємодії з іншими лікарськими засобами.
У процесі КВ нових фармакологічних засобів виділяють 4 взаємопов'язані фази:
1. Визначають безпеку лікарських засобів і встановлюють діапазон доз, що переносяться. Дослідження проводять на здорових чоловіках-добровольцях, у виняткових випадках – на хворих.
2. Визначають ефективність та переносимість лікарських засобів. Підбирається мінімальна ефективна доза, визначаються широта терапевтичної дії та підтримуюча доза. Дослідження проводять на хворих тієї нозології, на яку призначений досліджуваний препарат (50-300 осіб).
3. Уточнюють ефективність та безпеку препарату, його взаємодію з іншими лікарськими засобами порівняно зі стандартними методами лікування. Дослідження проводять на великій кількості пацієнтів (тисячі хворих), із залученням особливих груп хворих.
4. Післяреєстраційні маркетингові дослідження вивчають токсичні дії препарату при тривалому прийомі, виявляють рідкісні побічні ефекти. У дослідження можуть бути включені різні групи хворих – за віком, за новими показаннями.
Види клінічних досліджень:
Відкрите, коли всі учасники випробувань знають, який препарат одержує хворий;
Просте сліпе - хворий не знає, а дослідник знає, яке лікування було призначено;
У подвійному «сліпому» – ні штат дослідників, ні хворий не знають, чи отримує він препарат чи плацебо;
Потрійне "сліпе" - ні штат дослідників, ні перевіряючий, ні хворий не знають, яким препаратом він лікується.
Одним із різновидів КІ є дослідження на біоеквівалентність. Це основний вид контролю відтворених лікарських засобів, що не відрізняються лікарською формою та вмістом діючих речовин від відповідних оригіналів. Дослідження біоеквівалентності дозволяють зробити обґрунтовані
висновки про якість порівнюваних препаратів за меншим обсягом первинної інформації та у більш стислий термін. Проводяться здебільшого на здорових добровольцях.
На території Росії здійснюються клінічні дослідження всіх фаз. Більшість міжнародних клінічних досліджень та досліджень зарубіжних лікарських засобів відноситься до 3-ї фази, а у разі клінічних досліджень вітчизняних лікарських засобів значну їх частину становлять дослідження 4-ї фази.
У Росії за останні десять років склався спеціалізований ринок клінічних досліджень.Він добре структурований, тут працюють висококваліфіковані професіонали - лікарі-дослідники, науковці, організатори, менеджери та ін. статистики.
За період з жовтня 1998 р. до 1 січня 2005 р. було подано документи з проханням дозволити 1840 клінічних досліджень. У 1998-1999 роках. вітчизняні компанії становили вкрай незначну частку заявників, але починаючи з 2000 р. їхня роль помітно зросла: 2001 р. було 42%, 2002 р. - вже 63% заявників, 2003 р. - 45,5%. Серед зарубіжних країн-заявників є Швейцарія, США, Бельгія, Великобританія.
Об'єктом вивчення клінічних досліджень є лікарські засоби як вітчизняного, так і зарубіжного виробництва, сфера застосування яких зачіпає практично всі відомі розділи медицини. Найбільша кількість лікарських засобів застосовується для лікування серцево-судинних та онкологічних захворювань. Далі йдуть такі галузі, як психіатрія та неврологія, гастроентерологія, інфекційні хвороби.
Однією з тенденцій у розвитку сектора клінічних випробувань у нашій країні слід визнати швидке зростання числа КІ на біоеквівалентність препаратів-дженериків. Очевидно, що це відповідає особливостям російського фармацевтичного ринку: як відомо, він являє собою ринок відтворених препаратів.
Проведення клінічних випробувань у Росії регулюєтьсяКонституцією Російської Федерації,яка говорить, що «... ніхто не
може бути без добровільної згоди підданий медичним, науковим та іншим дослідам».
Деякі статті Федерального закону "Основи законодавства РФ про охорону здоров'я громадян"(від 22.07.1993 № 5487-1) визначають основи проведення КВ. Так, у статті 43 зазначено, що не дозволені до застосування, але перебувають на розгляді в установленому порядку лікарські засоби, можуть використовуватися на користь лікування пацієнта тільки після отримання його добровільної письмової згоди.
Федеральний Закон «Про лікарські засоби»№ 86-ФЗ має окремий розділ IX «Розробка, доклінічні та клінічні дослідження лікарських засобів» (статті 37-41). Тут зазначено порядок прийняття рішення про проведення КВ ЛЗ, правові основи проведення КВ та питання фінансування КВ, порядок їх проведення, права пацієнтів, що беруть участь у КВ.
Клінічні дослідження проводяться відповідно до Стандарту галузі ОСТ 42-511-99 «Правила проведення якісних клінічних випробувань у Російській Федерації»(затверджено МОЗ Росії 29 грудня 1998 р.) (Good Clinical Practice - GCP). Правила проведення якісних клінічних випробувань у Російській Федерації є етичний і науковий стандарт якості планування та проведення досліджень на людині, а також документального оформлення та подання їх результатів. Дотримання цих правил є гарантією достовірності результатів клінічних випробувань, безпеки, охорони прав та здоров'я піддослідних відповідно до основоположних принципів Гельсінської декларації. Вимоги цих Правил повинні дотримуватися під час проведення клінічних випробувань лікарських засобів, результати яких планується подати до дозвільних інстанцій.
GCP встановлюють вимоги до планування, проведення, документального оформлення та контролю клінічних досліджень, покликаних гарантувати захист прав, безпеку та охорону здоров'я осіб, які у них беруть участь, при проведенні яких не можна виключити небажаного впливу на безпеку та здоров'я людини, а також забезпечити достовірність та точність одержуваної у ході дослідження інформації. Правила є обов'язковими для виконання всіма учасниками клінічних досліджень лікарських засобів на території Російської Федерації.
З метою вдосконалення методичних основ проведення досліджень біоеквівалентності лікарських засобів, що є основним видом медико-біологічного контролю відтворених лікарських засобів, Міністерством охорони здоров'я та соціального розвитку Російської Федерації 10.08.2004 р. затверджено методичні вказівки "Проведення якісних клінічних досліджень біоеквівалентності лікарських засобів".
Згідно з нормативними документами, КІ ЛЗ проводятьсяу закладах охорони здоров'я, акредитованих федеральним органом виконавчої влади, до компетенції якого входить здійснення державного контролю та нагляду у сфері обігу лікарських засобів; він же складає та публікує перелік закладів охорони здоров'я, які мають право проводити клінічні дослідження лікарських засобів.
Правову основу проведення КВ ЛЗскладають рішення федерального органу виконавчої, до компетенції якого входить здійснення державного контролю та нагляду у сфері обігу лікарських засобів, про проведення КІ лікарського засобу та договір про його проведення. Рішення про проведення КВ ЛЗ приймається Федеральною службою з нагляду у сфері охорони здоров'я та соціального розвитку Російської Федерації відповідно до закону «Про лікарські засоби» та на підставі заяви, позитивного висновку комітету з етики при федеральному органі контролю якості лікарських засобів, звіту та висновку про доклінічні дослідженнях та інструкції з медичного застосування лікарського засобу.
За федерального органу контролю якості лікарських засобів створено Комітет з етики. Установа охорони здоров'я не розпочинає дослідження, допоки Комітет з етики не схвалить (в письмовому вигляді) форму письмової поінформованої згоди та інші матеріали, що надаються випробуваному або його законному представнику. Форма поінформованої згоди та інші матеріали можуть переглядатися протягом дослідження, якщо відкриваються обставини, здатні вплинути на згоду піддослідного. Нова редакція вищезазначеної документації в обов'язковому порядку проходить схвалення Комітету з етики, а факт доведення її до випробуваного має бути документально підтверджено.
Вперше у світовій практиці державний контроль за проведенням клінічних досліджень та дотриманням прав учасників експерименту було розроблено та здійснено у Пруссії. 29 жовтня 1900 р. Міністерство охорони здоров'я зобов'язало університетські клініки проводити клінічні експерименти за обов'язкової умови попереднього отримання пацієнтів письмової згоди. У 1930-ті роки. щодо прав людини ситуація у світі кардинально змінилася. У концтаборах для військовополонених у Німеччині, Японії експерименти на людях проводилися настільки масштабно, що згодом у кожному концтаборі навіть визначилася своя спеціалізація з медичних експериментів. Лише у 1947 р. міжнародний Військовий Трибунал повернувся до проблеми захисту прав людей, які беруть участь у проведенні клінічних досліджень. У процесі його роботи було вироблено перший міжнародний кодекс «Звід правил проведення експериментів на людях»,так званий Нюрнберзький кодекс.
У 1949 р. у Лондоні був прийнятий Міжнародний Кодекс Медичної Етики, який проголосив тезу про те, що «лікар повинен діяти лише на користь пацієнта, надаючи медичну допомогу, яка має покращувати фізичний та розумовий стан пацієнта», а Женевська конвенція Всесвітньої Асоціації Лікарів (1948) -1949 рр.), визначила обов'язок лікаря словами: «піклування про здоров'я мого пацієнта є моїм першим завданням».
Поворотним моментом у становленні етичної основи клінічних випробувань стало прийняття 18-ї Генеральної Асамблеєю Всесвітньої медичної асоціації в Гельсінкі в червні 1964 р. Гельсінської деклараціїВсесвітньої медичної асоціації, яка увібрала в себе весь світовий досвід етичного змісту біомедичних досліджень. З того часу Декларація неодноразово переглядалася, востаннє - в Единбурзі (Шотландія) у жовтні 2000 року.
У Гельсінській декларації записано, що біомедичні дослідження за участю людей повинні відповідати загальноприйнятим науковим принципам і ґрунтуватися на адекватно проведених лабораторних дослідженнях та експериментах на тваринах, а також достатньому знанні наукової літератури. Вони мають проводитися кваліфікованим персоналом під наглядом досвідченого лікаря. У всіх випадках відповідальність за пацієнта несе лікар, але не сам пацієнт, незважаючи на дану їм поінформовану згоду.
При будь-яких дослідженнях за участю людей як суб'єктів кожен потенційний учасник повинен бути відповідним чином поінформований про цілі, методи, очікувану користь дослідження та про пов'язані з ним ризики та незручності. Люди повинні бути поінформовані про те, що вони мають право утриматися від участі у дослідженні та можуть у будь-який час після його початку анулювати свою згоду та відмовитись від продовження дослідження. Потім лікар повинен отримати від суб'єкта вільно дану інформовану згоду письмово.
Іншим важливим документом, який визначає етичні норми проведення клінічних випробувань, стало «Міжнародний посібник з етики біомедичних досліджень із залученням людини»,ухвалена Радою Міжнародних організацій з медичної науки (CIOMS) (Женева, 1993 р.), яка містить рекомендації для дослідників, спонсорів, представників охорони здоров'я та етичних комітетів про те, як впроваджувати етичні стандарти в галузь медичних досліджень, а також етичні принципи щодо всіх осіб, включаючи пацієнтів, які беруть участь у клінічних дослідженнях.
Гельсінська декларація та «Міжнародний посібник з етики біомедичних досліджень із залученням людини» показують, як фундаментальні етичні принципи можуть бути ефективно застосовані в практиці медичних досліджень у всьому світі, при цьому враховуються різні особливості культур, релігій, традицій, соціальних та економічних умов, законів, адміністративних систем та інших ситуацій, які можуть мати місце у країнах з обмеженими ресурсами.
19 листопада 1996 р. Парламентською асамблеєю Ради Європи було прийнято «Конвенція про захист прав людини та людської гідності у зв'язку із застосуванням біології та медицини».Норми, закладені у Конвенції, мають як силу морального призову - кожна держава, що приєдналася до неї, бере він зобов'язання втілити «основні її становища у національному законодавстві». Відповідно до положень цієї Конвенції інтереси та благо окремої людини переважають інтереси суспільства і науки. Будь-яке медичне втручання, включаючи втручання з дослідницькими цілями, повинно здійснюватися відповідно до професійних вимог та стандартів. Випробовуваний зобов'язаний заздалегідь отримати відповідну інформацію про мету та характер втручання, а також про
його наслідки та ризики; його згода має бути добровільною. Медичне втручання щодо особи, яка не здатна дати на це згоду, може здійснюватися виключно у безпосередніх її інтересах. 25 січня 2005 р. було прийнято Додатковий протокол до Конвенції, що стосується біомедичних досліджень.
Для забезпечення гарантій дотримання прав випробуваних на даний час міжнародне співтовариство виробило ефективну систему громадського та державного контролю за забезпеченням прав та інтересів суб'єктів досліджень та етичності клінічних випробувань. Однією з основних ланок системи громадського контролю є діяльність незалежних етичних комітетів(ЕК).
Комітети з етики є сьогодні структурами, у полі зору яких схрещуються наукові інтереси, медичні факти та норми моралі та права. Комітети з етики здійснюють функції експертизи, консультування, рекомендацій, спонукання, оцінки, орієнтування у моральних та правових питаннях КІ. Етичні комітети відіграють вирішальну роль у визначенні того, що дослідження безпечне, проводиться сумлінно, що дотримуються права пацієнтів, які в ньому беруть участь, іншими словами, ці комітети гарантують суспільству, що кожне клінічне дослідження, що проводиться, відповідає етичним стандартам.
ЕК мають бути незалежні від дослідників і повинні отримувати матеріальну вигоду від проведених досліджень. Дослідник повинен отримати пораду, сприятливе відкликання або дозвіл комітету перед початком роботи. Комітет здійснює подальший контроль, може вносити поправки до протоколу та моніторувати хід та результати дослідження. Етичні комітети повинні мати повноваження заборонити проведення дослідження, припинити його проведення або просто відхилити або припинити дію виданого дозволу.
Основними засадами діяльності комітетів з етикипри здійсненні етичної експертизи КІ є незалежність, компетентність, відкритість, плюралізм, а також об'єктивність, конфіденційність, колегіальність.
ЕК мають бути незалежними від органів, що приймають рішення про проведення КВ, у тому числі від державних органів. Неодмінною умовою компетентності Комітету є висока кваліфікація та чітка робота його протокольної групи (або
секретаріату). Відкритість діяльності комітету з етики забезпечується прозорістю засад його роботи, регламенту тощо. Стандартні операційні процедури мають бути відкриті всім охочих ознайомитися з ними. Плюралізм комітету з етики гарантується різнорідністю професій, віку, статі, конфесій його членів. У процесі експертизи мають враховуватися права всіх учасників дослідження, зокрема не лише пацієнтів, а й лікарів. Дотримання конфіденційності потрібне щодо матеріалів КІ, осіб, які беруть участь у ньому.
Незалежний етичний комітет зазвичай створюється під егідою національного чи місцевого департаментів охорони здоров'я, на базі медичних установ чи інших національних, регіональних, місцевих представницьких органів – як громадське об'єднання без утворення юридичної особи.
Основними цілями роботи етичного комітетує захист прав та інтересів піддослідних та дослідників; неупереджена етична оцінка клінічних та доклінічних досліджень (випробувань); забезпечення проведення якісних клінічних та доклінічних досліджень (випробувань) відповідно до міжнародних норм; забезпечення впевненості громадськості у тому, що будуть гарантовані та дотримані всі етичні засади.
Для виконання зазначених цілей етичний комітет повинен вирішувати такі завдання: незалежно та об'єктивно оцінювати безпеку та недоторканність прав людини стосовно до випробуваних як на стадії планування, так і на стадії проведення дослідження (випробування); оцінювати відповідність дослідження гуманістичним та етичним нормам, доцільність проведення кожного дослідження (випробування), відповідність дослідників, технічних засобів, протоколу (програми) проведення дослідження, підбору суб'єктів дослідження, якості рандомізації правил проведення якісних клінічних випробувань; здійснювати контроль за дотриманням стандартів якості проведення клінічних досліджень для забезпечення достовірності та повноти даних.
Оцінка співвідношення ризику та користіє найважливішим етичним рішенням, яке приймає ЕК під час експертизи дослідницьких проектів. Для визначення розумності ризиків стосовно користі треба врахувати ряд факторів, причому кожен випадок слід розглядати індивідуально, приймати
до уваги особливості випробуваних, що у дослідженні (діти, вагітні жінки, невиліковні хворі).
Для проведення оцінки ризиків та очікуваної користі ЕК повинен переконатися, що:
Необхідні дані неможливо отримати без залучення до дослідження людей;
Дослідження раціонально сплановано з урахуванням мінімізації дискомфорту та інвазивних процедур для випробуваних;
Дослідження служить отриманню важливих результатів, спрямованих на вдосконалення діагностики та лікування або сприяють узагальненню та систематизації даних про захворювання;
Дослідження базується на результатах лабораторних даних та експериментів на тваринах, поглибленому знанні історії проблеми, а очікувані результати лише підтвердять його обґрунтованість;
Очікувана користь від дослідження перевищує потенційний ризик, а потенційний ризик мінімальний, тобто. не більшим, ніж при виконанні звичайних лікувальних та діагностичних процедур при даній патології;
Дослідник має достатню інформацію про передбачуваність будь-яких можливих несприятливих наслідків дослідження;
Випробуваним та їхнім законним представникам надано всю інформацію, необхідну отримання їх усвідомленого і добровільного згоди.
Клінічні дослідження повинні здійснюватися відповідно до положень міжнародних та державних законодавчих документів, що гарантують захист прав випробуваного.
Записані в Конвенції про захист прав людини положення стоять на варті гідності та індивідуальної цілісності людини та гарантують кожному без винятку дотримання недоторканності особистості та інших прав та основних свобод у зв'язку із застосуванням досягнень біології та медицини, у тому числі в галузі трансплантології, генетики, психіатрії та ін.
Жодне дослідження на людині не може бути проведене без одночасного дотримання всіх перерахованих умов:
Немає альтернативних методів дослідження, порівнянних за своєю ефективності;
Ризик, якому може бути підданий випробуваний, не перевищує потенційної вигоди від цього дослідження;
Проект запропонованого дослідження було затверджено компетентним органом після проведення незалежної експертизи наукової обґрунтованості проведення даного дослідження, включаючи важливість його мети та багатостороннього розгляду його прийнятності з етичної точки зору;
Особа, яка виступає як випробуваний, поінформована про наявні в нього права та гарантії, передбачені законом;
Отримано письмову поінформовану згоду на проведення експерименту, яка може бути безперешкодно відкликана будь-якої миті.
В «Основах законодавства РФ про охорону здоров'я громадян» та у ФЗ «Про лікарські засоби» закріплено положення, що будь-яке біомедичне дослідження із залученням людини як об'єкт має проводитися тільки після отримання письмової згоди громадянина. Людина може бути змушений до участі у біомедичному дослідженні.
При отриманні згодина біомедичне дослідження громадянину має бути надана інформація:
1) про лікарський засіб та сутність його клінічних досліджень;
2) про очікувану ефективність, про безпеку лікарського засобу, ступінь ризику для пацієнта;
3) про дії пацієнта у разі непередбачених ефектів впливу лікарського засобу на стан його здоров'я;
4) про умови страхування здоров'я пацієнта.
Пацієнт має право відмовитися від участі у клінічних дослідженнях на будь-якій стадії їх проведення.
Інформація про дослідження має бути доведена до пацієнта у доступній та зрозумілій формі. Дослідник або його співробітник зобов'язані до отримання поінформованої згоди дати випробуваному або його представнику достатньо часу для ухвалення рішення про участь у дослідженні та надати можливість отримати детальну інформацію про випробування.
Інформована згода (згода поінформованого пацієнта) гарантує, що майбутні піддослідні розуміють характер дослідження та можуть зі знанням справи та добровільно прийняти рішення
про свою участь чи неучасть. Ця гарантія захищає всі сторони: як випробуваного, до самостійності якого проявляється повага, і дослідника, який інакше входить у суперечність із законом. Інформована згода є однією з головних етичних вимог до досліджень за участю людей. Воно відбиває фундаментальний принцип поваги до особистості. До елементів поінформованої згоди належать повне розкриття інформації, адекватне розуміння та добровільний вибір. До медичних досліджень можуть залучатись різні групи населення, але забороняється проведення клінічних досліджень лікарських засобів на:
1) неповнолітніх, які мають батьків;
2) вагітних жінок, за винятком випадків, коли проводяться клінічні дослідження лікарських засобів, призначених для вагітних жінок і коли повністю виключено ризик заподіяння шкоди вагітній жінці та плоду;
3) особах, які відбувають покарання у місцях позбавлення волі, а також на тих, хто перебуває під вартою у слідчих ізоляторах, без їх письмової поінформованої згоди.
Допускаються клінічні дослідження лікарських засобів на неповнолітніх тільки тоді, коли досліджуваний засіб призначається виключно для лікування дитячих хвороб або метою клінічних випробувань є отримання даних про найкраще дозування препарату для лікування неповнолітніх. У разі клінічним дослідженням на дітях мають передувати аналогічні випробування на повнолітніх. У ст. 43 Основ законодавства РФ «про охорону здоров'я громадян» зазначено: «Не дозволені до застосування, але перебувають на розгляді в установленому порядку методи діагностики, лікування та лікарські засоби можуть використовуватися для лікування осіб, які не досягли віку 15 років, лише за безпосередньої загрози їх життю та за письмовою згодою їх законних представників». Інформація про дослідження повинна бути передана дітям мовою, доступною для їхнього розуміння з урахуванням віку. Підписана поінформована згода може бути отримана від дітей, які досягли відповідного віку (з 14 років, що визначається законодавством та етичними комітетами).
Допускаються клінічні випробування препаратів, призначених для лікування психічних захворювань, на особах із психічними захворюваннями та визнаних недієздатними в порядку,
встановленому Законом Російської Федерації №3185-1 від 02.07.1992 р. «Про психіатричну допомогу та гарантії прав громадян при її наданні». Клінічні дослідження лікарських засобів у разі проводяться за наявності письмової згоди законних представників зазначених осіб.
Глава 3. КЛІНІЧНІ ДОСЛІДЖЕННЯ ЛІКАРСЬКИХ ЗАСОБІВ
Появі нових ЛЗ передує тривалий цикл досліджень, завдання яких – довести ефективність та безпеку нового препарату. Принципи доклінічних досліджень на лабораторних тварин були розроблені оптимально, але в 1930-х роках стало ясно, що результати, отримані в експерименті на тваринах, не можна безпосередньо переносити на людину.
Перші клінічні дослідження у людини були проведені на початку 1930-х років (1931 р. - перше рандомізоване сліпе дослідження санокризину ** 3, 1933 р. - перше плацебоконтрольоване дослідження у хворих зі стенокардією). В даний час у всьому світі проведено кілька сотень тисяч клінічних досліджень (по 30 000-40 000 на рік). Появі кожного нового препарату передує в середньому 80 різних досліджень за участю понад 5000 пацієнтів. Це суттєво подовжує період розробки нових препаратів (у середньому 14,9 року) і потребує значних витрат: лише на клінічні дослідження компанії-виробники витрачають у середньому 900 млн дол. Проте лише проведення клінічних досліджень гарантує отримання точної та достовірної інформації про безпеку та ефективність нового препарату.
Відповідно до міжнародного керівництва з Якісної клінічної практики (міжнародний стандарт проведення клінічних досліджень: ICH/GCP), під клінічним дослідженнямрозуміють «вивчення безпеки та/або ефективності досліджуваного препарату у людини, спрямоване на виявлення або підтвердження клінічних, бажаних фармакодинамічних властивостей досліджуваного препарату та/або проведене з метою виявлення його побічних ефектів та/або з метою вивчення його всмоктування, розподілу, біотрансформації та виведення» .
Ціль клінічного дослідження- отримання достовірних даних про ефективність та безпеку препарату, не піддаючи
при цьому пацієнтів (суб'єктів дослідження) є необґрунтованим ризиком. Більш конкретно дослідження може ставити за мету вивчення фармакологічної дії препарату на людину, встановлення терапевтичної (лікувальної) ефективності або підтвердження ефективності в порівнянні з іншими ЛЗ, а також визначення терапевтичного застосування - тієї ніші, яку може займати даний препарат у сучасній фармакотерапії. Крім того, дослідження може бути етапом підготовки препарату до реєстрації, сприяти просування ринку вже зареєстрованого препарату або бути інструментом вирішення наукових проблем.
3.1. СТАНДАРТИ В ОБЛАСТІ КЛІНІЧНИХ ДОСЛІДЖЕНЬ
До появи єдиних стандартів клінічних досліджень хворі, які отримували нові ЛЗ, часто наражалися на серйозний ризик, пов'язаний з прийомом недостатньо ефективних і небезпечних препаратів. Наприклад, на початку ХХ ст. у ряді країн героїн використовували як засіб для лікування кашлю; в 1937 р. у США кілька десятків дітей загинули після прийому сиропу парацетамолу, до складу якого входив токсичний етиленглі-кіль *; а у 1960-х роках у Німеччині та Великобританії у жінок, які приймали під час вагітності талідомід*, народилося близько 10 000 дітей із серйозними аномаліями розвитку кінцівок. Неправильне планування досліджень, помилки в аналізі результатів та відверті фальсифікації стали причиною низки інших гуманітарних катастроф, що поставило питання про законодавчий захист інтересів хворих, які беруть участь у дослідженнях, та потенційних споживачів ЛЗ.
Сьогодні потенційний ризик призначення нових ЛЗ істотно нижчий, оскільки державні органи, що дають своє схвалення на їх використання, мають можливість оцінити результати застосування нового препарату у тисяч хворих під час клінічних досліджень, виконаних за єдиним стандартом.
В даний час всі клінічні дослідження проводять за єдиним міжнародним стандартом, що отримав назву GCP , який був розроблений Адміністрацією з контролю за лікарем-
ними засобами та харчовими продукціями уряду США, ВООЗ та Євросоюзом у 1980-1990-х роках. Стандарт GCP регламентує планування та проведення клінічних досліджень, а також передбачає багатоступеневий контроль безпеки пацієнтів та точність отриманих даних.
У стандарті GCP враховано етичні вимоги до проведення наукових досліджень за участю людини, сформульовані Гельсінською декларацією Всесвітньої медичної асоціації«Рекомендації для лікарів, які займаються біомедичними дослідженнями за участю людей». Зокрема, участь у клінічних дослідженнях може бути лише добровільною, в ході досліджень хворі не повинні отримувати грошову винагороду. Підписуючи свою згоду стати учасником дослідження, пацієнт отримує точну та детальну інформацію про можливий ризик для свого здоров'я. Крім того, хворий може припинити свою участь у дослідженні будь-якої миті без пояснення причин.
Клінічна фармакологія, що вивчає фармакокінетику та фар-макодинаміку ЛЗ безпосередньо у хворої людини, мала велике значення при створенні стандартів GCP та всієї сучасної концепції клінічних досліджень ЛЗ.
Положення міжнародного стандарту ICH GCP знайшли відображення у Федеральний закон «Про обіг лікарських засобів»(№ 61-ФЗ від 12.04.2010 р.) та Державний стандарт «Належна клінічна практика»(ГОСТ Р 52379-2005), яким виконуються клінічні дослідження ЛЗ нашій країні. Таким чином, існує юридична підстава для взаємного визнання результатів клінічних досліджень різними країнами, а також проведення великих міжнародних клінічних досліджень.
3.2. ПЛАНУВАННЯ І ПРОВЕДЕННЯ КЛІНІЧНИХ ДОСЛІДЖЕНЬ
Планування клінічного дослідження включає кілька етапів.
Визначення дослідницького питання. Наприклад, чи справді препарат X достовірно знижує АТ у хворих на АГ або чи справді препарат X здатний знижувати АТ більш ефективно, ніж препарат Y. Дослідження, як правило, має один основний і кілька додаткових дослідницьких
питань, наприклад: чи може препарат Z знижувати смертність хворих на АГ (основне питання), як препарат Z при цьому впливає на частоту госпіталізацій, яка частина хворих з помірною гіпертензією, у яких препарат Z здатний надійно контролювати рівень АТ (додаткові питання). Дослідницьке питання відображає припущення, з якого виходять дослідники (Гіпотезу дослідження);у нашому прикладі гіпотеза полягає в тому, що препарат Z, маючи здатність знижувати АТ, може зменшувати ризик пов'язаних з артеріальною гіпертензією ускладнень, захворювань і, отже, дозволяє зменшити частоту летальних наслідків.
Вибір дослідження дизайну. Дослідження може включати кілька груп порівняння (препарат А і плацебо або препарат А і препарат B). Дослідження, в яких немає групи порівняння, не дають змоги отримати достовірну інформацію про дію ЛЗ, і нині такі дослідження практично не проводять.
Визначення обсягу вибірки. Автори протоколу повинні передбачити, яка саме кількість хворих буде потрібна для доказу вихідної гіпотези (величину обсягу вибірки розраховують математично на підставі законів статистики). У дослідження можна включати від кількох десятків (у тому випадку, коли ефект препарату значно виражений) до 30 000-50 000 пацієнтів (якщо ефект препарату менш виражений).
Визначення тривалості дослідження. Тривалість дослідження залежить від часу настання ефекту. Наприклад, бронхолітики покращують стан хворих на бронхіальну астму вже через кілька хвилин після їх прийому, а зареєструвати позитивну дію інгаляційних глюкокортикоїдів у цих хворих можна лише через кілька тижнів. Крім того, низка досліджень потребує спостереження за порівняно рідкісними подіями: якщо очікується, що досліджуваний препарат здатний знизити кількість загострень захворювання, для підтвердження цього ефекту необхідне тривале спостереження. У сучасних дослідженнях терміни спостереження становлять від кількох годин до 5-7 років.
Вибір популяції хворих. Для потрапляння у дослідження хворих із певними характеристиками розробники створюють чіткі критерії. Вони передбачають вік, стать, тривалість та тяжкість захворювання, характер попереднього
лікування, супутні захворювання, які можуть вплинути на оцінку ефекту ЛЗ. Критерії включення повинні забезпечити однорідність пацієнтів. Наприклад, якщо до дослідження з АГ будуть одночасно включені хворі з легкою (прикордонною) гіпертензією та хворі з дуже високими значеннями АТ, досліджуваний препарат впливатиме на цих пацієнтів по-різному, що ускладнить отримання надійних результатів. Крім того, у дослідження зазвичай не включають вагітних та осіб з тяжкими захворюваннями, що негативно впливають на загальний стан та прогноз хворого.
Методи оцінки ефективності лікування. Розробники повинні вибрати індикатори ефективності препарату, у нашому прикладі слід уточнити, як саме буде оцінено гіпотензивний ефект шляхом одноразового вимірювання АТ; шляхом обчислення середньодобової величини АТ; ефективність лікування буде оцінена за впливом на якість життя хворого або за здатністю ЛЗ запобігати проявам ускладнень АГ.
Методи оцінки безпеки. Слід передбачити заходи щодо оцінки безпеки лікування та способи реєстрації НЛР досліджуваних препаратів.
Етап планування завершується написанням протоколу - основного документа, що передбачає порядок проведення дослідження та всі дослідницькі процедури. Таким чином, протокол дослідження«описує завдання, методологію, статистичні аспекти та організацію дослідження». Протокол надають для ознайомлення до державних регуляторних органів та незалежного етичного комітету, без схвалення яких не можна приступити до виконання дослідження. Внутрішній (моніторинг) та зовнішній (аудит) контроль за проведенням дослідження оцінює насамперед відповідність дій дослідників процедурі, описаній у протоколі.
Включення пацієнтів у дослідження- Суто добровільне. Обов'язкова умова включення – ознайомлення пацієнта з можливим ризиком та користю, яку він може отримати з участі у дослідженні, а також підписання ним поінформованої згоди.Правила ICH GCP не допускають використання матеріальних стимулів для залучення хворих до участі у дослідженні (виняток роблять для здорових волонтерів, які залучаються для дослідження фармакокінетики або біоеквівалентності ЛЗ). Хворий повинен відповідати критеріям включення/виключення. Зазвичай
не допускають участь у дослідженнях вагітних, матерів-годувальниць, хворих, у яких може бути змінена фармакокінетика досліджуваного препарату, хворих на алкоголізм або наркоманію. Неприпустиме включення до дослідження недієздатних пацієнтів без згоди піклувальників, військовослужбовців, ув'язнених, осіб з алергією на досліджуваний препарат чи хворих, які одночасно беруть участь в іншому дослідженні. Хворий має право припинити свою участь у дослідженні будь-якої миті без пояснення причин.
Дизайн дослідження.Дослідження, в ході яких усі пацієнти отримують однакове лікування, в даний час практично не проводять через низьку доказовість результатів. Найбільш поширене порівняльне дослідження у паралельних групах (група втручання та група контролю). Для контролю можна використовувати плацебо (плацебоконтрольоване дослідження) чи інший активний препарат.
Дослідження з порівняльним дизайном вимагають рандомізації- розподілу учасників на дослідну та контрольну групи випадковим чином, що дозволяє звести до мінімуму систематичну помилку та упередженість. Дослідник в принципі може отримати доступ до інформації про те, яке ЛЗ отримує хворий (це може знадобитися у разі серйозних небажаних реакцій), але в цьому випадку пацієнта необхідно виключити з дослідження.
Індивідуальна реєстраційна картка.Під індивідуальною реєстраційною карткою розуміють «друкований, оптичний або електронний документ, створений для реєстрації всієї інформації, що вимагається в протоколі, про кожного суб'єкта дослідження». З індивідуальної реєстраційної карти створюють базу даних дослідження щодо статистичної обробки результатів.
3.3. ФАЗИ КЛІНІЧНОГО ДОСЛІДЖЕННЯ ЛІКАРСЬКИХ ЗАСОБІВ
І виробник, і суспільство зацікавлені в отриманні якомога більш точної та повної інформації про клінічну фармакологію, терапевтичну ефективність та безпеку нового ЛЗ у ході досліджень, що передують реєстрації. Підготовка
реєстраційне досьє неможливе без відповіді на ці питання. Через цю реєстрацію нового ЛЗ передують кілька десятків різних досліджень, причому з кожним роком збільшується як кількість досліджень, і кількість їх учасників, а загальний цикл досліджень нового ЛЗ зазвичай перевищує 10 років. Таким чином, розробка нових ЛЗ можлива лише у великих фармацевтичних компаніях, а загальна вартість дослідницького проекту в середньому перевищує 900 млн. дол.
Перші, доклінічні дослідження починаються невдовзі після синтезу нової, потенційно ефективної молекули. Їх суть полягає у перевірці гіпотези про передбачувану фармакологічну дію нової сполуки. Паралельно вивчають токсичність сполуки, її онкогенну та тератогенну дію. Всі ці дослідження виконують на лабораторних тваринах, а їхня загальна тривалість становить 5-6 років. Внаслідок такої роботи з 5-10 тис. нових сполук відбирають приблизно 250.
Власне клінічні дослідження умовно поділяють на чотири періоди чи фази.
І фазу клінічних досліджень,зазвичай проводять на 28-30 здорових добровольцях. Мета цього етапу - отримання відомостей про переносимість, фармакокінетику та фармакодинаміку нового ЛЗ, уточнення режиму дозування та отримання даних щодо безпеки препарату. Вивчення терапевтичної дії препарату у цій фазі необов'язкове, оскільки у здорових добровольців ряд клінічно важливих властивостей нового ЛЗ зазвичай не спостерігають.
Дослідження I фази починають з вивчення безпеки та фармакокінетики одноразової дози, при виборі якої використовують дані, отримані на біологічних моделях. Надалі вивчають фармакокінетику препарату при багаторазовому призначенні, екскрецію та метаболізм нового ЛЗ (порядок кінетичних процесів), його розподіл у рідинах, тканинах організму, фармакодинаміку. Зазвичай усі ці дослідження проводять для різних доз, лікарських форм та шляхів введення. У ході I фази досліджень можна також оцінювати вплив на фармакокінетику та фармакодинаміку нового препарату інших ЛЗ, функціонального стану організму, прийому їжі тощо.
Важливою метою I фази клінічних випробувань вважають виявлення потенційної токсичності та НЛР, але ці дослідження нетривалі і їх проводять у обмеженої кількості учасників, отже, в ході цієї фази вдається виявити лише найбільше
часті та виражені небажані явища, пов'язані із застосуванням нового ЛЗ.
У ряді випадків (онкологічні препарати, препарати для терапії ВІЛ-інфекції) дослідження І фази можна проводити у хворих. Це дозволяє прискорити створення нового препарату і не наражати добровольців на необґрунтований ризик, хоча такий підхід можна розглядати швидше як виняток.
Дослідження I фазидозволяють:
Оцінити переносимість та безпеку нового препарату;
У ряді випадків отримати уявлення про його фармакокінетику (у здорових людей, що природно має обмежене значення);
Визначити основні фармакокінетичні константи (C max ,
C1);
Порівняти фармакокінетику нового препарату при використанні різних лікарських форм, шляхів та способів призначення.
Дослідження ІІ фази- Перші дослідження у хворих. Обсяг цих досліджень значно більший, ніж у I фазі: 100-200 пацієнтів (іноді до 500). У II фазі уточнюють ефективність та безпеку нового препарату, а також діапазон доз для лікування хворих. Ці дослідження дають інформацію переважно про фармакодинаміку нового ЛЗ. Обов'язковими умовами проведення досліджень ІІ фази вважають порівняльний дизайн та включення контрольної групи (що нехарактерно для досліджень І фази).
Дослідження ІІІ фазипланують для великої кількості пацієнтів (до 10 000 чоловік і більше), а умови їхнього проведення максимально наближені до звичайних умов лікування певних захворювань. Дослідження цієї фази (як правило, це кілька паралельно або послідовно проведених досліджень) великі (повномасштабні), рандомізовані та порівняльні. Предметом вивчення стає як фармакодинаміка нового ЛЗ, а й його клінічна ефективність 1 .
1 Наприклад, мета дослідження нового гіпотензивного препарату в І-ІІ фазі – довести його здатність знижувати АТ, а при дослідженні ІІІ фази метою стає вивчення впливу ЛЗ на АГ. У разі разом із зниженням АТ з'являються інші точки оцінки ефекту, зокрема зниження смертності від серцево-судинних захворювань, профілактика ускладнень АГ, підвищення якості життя хворих тощо.
У III фазі досліджень препарат порівнюють з ефективності та безпеки з плацебо (плацебоконтрольоване дослідження) або/і з іншим маркерним препаратом (ЛЗ, що зазвичай використовується в даній клінічній ситуації і має добре відомі лікувальні властивості).
Подання компанією-розробником заявки на реєстрацію ЛЗ не означає завершення досліджень. Дослідження III фази, виконані до подачі заявки, називають дослідженнями Ша фази, а після подачі заявки – Шб фази. Останні проводять для отримання більш повної інформації про клінічну та фармакоекономічну ефективність ЛЗ. Такі дослідження можуть розширити показання до призначення нового ЛЗ. Ініціатором додаткових досліджень можуть стати державні органи, які відповідають за процес реєстрації, якщо результати попередніх досліджень не дозволяють однозначно висловитися про властивості та безпеку нового ЛЗ.
Результати досліджень III фази стають визначальними після ухвалення рішення про реєстрацію нового ЛЗ. Таке рішення може бути прийняте, якщо препарат:
Більш ефективний, ніж відомі ЛЗ аналогічної дії;
Має ефекти, які не властиві існуючим препаратам;
Має більш вигідну лікарську форму;
Більш вигідний у фармакоекономічному відношенні або дозволяє використовувати простіші методи лікування;
Має переваги при сумісному застосуванні з іншими ЛЗ;
Має простіший спосіб застосування.
Дослідження IV фази.Конкуренція з новими препаратами змушує продовжувати дослідження та після реєстрації нового ЛЗ (постмаркетингові дослідження) для підтвердження ефективності препарату та його місця у фармакотерапії. Крім того, дослідження IV фази дозволяють відповісти на деякі питання, що виникають в ході застосування ЛЗ (оптимальна тривалість лікування, переваги та недоліки нового препарату в порівнянні з іншими, у тому числі новішими ЛЗ, особливості призначення у літніх, дітей, віддалені ефекти лікування, нові свідчення і т.д.).
Іноді дослідження IV фази проводять багато років після реєстрації ЛЗ. Прикладом подібних відстрочених на понад 60 років
Клінічні дослідження всіх фаз проводять в офіційно сертифікованих державними органами контролю 2 центрах (медичні центри, лікарні, поліклініки), які мають відповідне науково-діагностичне оснащення та можливість надання кваліфікованої медичної допомоги хворим на НЛР.
Дослідження біоеквівалентності.Більшість ЛЗ на фармацевтичному ринку – відтворені (генеричні) препарати. Фармакологічна дія та клінічна ефективність ЛЗ, що входять до складу цих препаратів, як правило, досить добре вивчені. Однак ефективність генериків може суттєво різнитися.
Реєстрація генеричних препаратів може бути спрощеною (за часом та за обсягом досліджень). Зробити суворо обґрунтований висновок щодо якості цих засобів дозволяють дослідження біоеквівалентності. У цих дослідженнях генеричний препарат порівнюють з оригінальним біодоступністю (порівнюють частки препарату, що досягають системного кровотоку, і швидкість, з якою цей процес відбувається). Якщо два ЛЗ мають однакову біодоступність - вони біоеквівалентні. При цьому припускають, що біоеквівалентні препарати мають однакову ефективність та безпеку 3 .
Біоеквівалентність вивчають на невеликій кількості здорових добровольців (20-30), при цьому використовують стандартні для дослідження фармакокінетики процедури (побудова фармакокінетичної кривої, дослідження величини AUC, T max , тах).
max max
1 Запропоновані в клінічну практику близько 100 років тому, ці препарати свого часу не проходили процес реєстрації та клінічних досліджень, що й потребувало їх різнобічних досліджень понад 60 років. Сучасна система реєстрації нових препаратів з'явилася в 60-х роках XX ст., отже, близько 30-40% застосовуваних сьогодні ЛЗ були переконливо досліджені. Їхнє місце у фармакотерапії може бути предметом дискусії. В англомовній літературі для цих препаратів застосовують термін «ліки-сироти», оскільки рідко вдається знайти джерела фінансування для досліджень таких ЛЗ.
2 У нашій країні - МОЗсоцрозвитку РФ.
3 Однак не можна стверджувати, що два фармацевтично еквівалентні препарати (з однаковою ефективністю та безпекою) завжди мають однакову фармако-кінетику та порівнянну біодоступність.
3.4. ЕТИЧНІ АСПЕКТИ КЛІНІЧНИХ
ДОСЛІДЖЕНЬ
Найважливіший принцип медичної етики було сформульовано майже 2500 років тому. У клятві Гіппократа говориться: «Я зобов'язуюсь робити все це відповідно до моїх можливостей і знань на користь хворому і утримуватися від усього, що може завдати йому шкоди». Вимоги медичної деонтології набувають особливого значення при проведенні клінічних досліджень ЛЗ через їх проведення на людях та торкання прав людини на здоров'я та життя. Отже, медико-юридичні та медико-деонтологічні проблеми мають велике значення у клінічній фармакології.
При проведенні клінічних досліджень ЛЗ (як нових, так і вже вивчених, але застосовуваних за новими показаннями) слід керуватися насамперед інтересами пацієнта. Дозвіл на проведення клінічних досліджень ЛЗ приймають компетентні органи (в РФ - МОЗ соціального розвитку Росії) після докладного вивчення сукупності даних, отриманих при доклінічному вивченні препарату. Однак, незалежно від дозволу державних інстанцій, дослідження також має отримати схвалення в комітеті з етики.
Етичну експертизу клінічних досліджень проводять відповідно до принципів Гельсінської декларації Всесвітньої медичної асоціації «Рекомендації для лікарів, які займаються біомедичними дослідженнями за участю людей» (вперше прийнята 18-ю Всесвітньою медичною асамблеєю в Гельсінкі в 1964 р. і потім була неодноразово переглянута).
У Гельсінській декларації є твердження, що метою біомедичних досліджень на людях мають бути покращення діагностичних, терапевтичних та профілактичних процедур, а також з'ясування етіології та патогенезу захворювань. Всесвітня медична асамблея підготувала рекомендації лікаря під час проведення клінічних досліджень.
Вимоги Гельсінської декларації було враховано у Федеральному законі РФ «Про обіг лікарських засобів». Зокрема, законодавчо підтверджено таке.
Участь пацієнтів у клінічних дослідженнях ЛЗ може бути лише добровільною.
Пацієнт дає письмову згоду на участь у клінічних дослідженнях ЛЗ.
Пацієнт повинен бути поінформований про характер дослідження та можливий ризик для свого здоров'я.
Пацієнт має право відмовитися від участі у клінічних дослідженнях ЛЗ на будь-якій стадії їх проведення.
За етичними вимогами неприпустимі клінічні дослідження ЛЗ щодо неповнолітніх (за винятком тих випадків, коли досліджуване ЛЗ призначене виключно для лікування дитячих хвороб) та вагітних. Заборонено проведення клінічних досліджень ЛЗ у неповнолітніх, які не мають батьків, у недієздатних осіб, ув'язнених, військовослужбовців тощо. Усі учасники клінічних досліджень мають бути застраховані.
Питаннями етичної експертизи клінічних досліджень нашій країні займається комітет з етики МОЗсоцразвития Росії, і навіть локальні комітети з етики при лікувальних і наукових медичних установах. Комітет з етики керується основними міжнародними принципами проведення клінічних досліджень, а також чинним законодавством та нормативно-правовими актами Російської Федерації.
3.5. ПРОЦЕДУРА РЕЄСТРАЦІЇ НОВИХ ЛІКАРСЬКИХ ЗАСОБІВ
Згідно з Федеральним законом «Про обіг лікарських засобів» (№61-ФЗ від 12.04.2010 р.), «ЛЗ можуть вироблятися, продаватися та застосовуватися на території Російської Федерації, якщо вони зареєстровані федеральним органом контролю якості ЛЗ». Державній реєстрації підлягають:
Нові ЛЗ;
Нові поєднання зареєстрованих раніше ЛЗ;
ЛЗ, зареєстровані раніше, але вироблені в інших лікарських формах або в новому дозуванні;
Генеричні препарати.
Державною реєстрацією ЛЗ займається МОЗ соціально-розвитку Росії, він же затверджує інструкцію щодо застосування ЛЗ, а зареєстроване ЛЗ заносять до державного реєстру.
Клінічна фармакологія та фармакотерапія: підручник. - 3-тє вид., перероб. та дод. / За ред. В. Г. Кукеса, А. К. Стародубцева. – 2012. – 840 с.: іл.
1. Клінічні дослідження лікарських засобів для медичного застосування, у тому числі міжнародні багатоцентрові, багатоцентрові, післяреєстраційні, проводяться в одній або кількох медичних організаціях відповідно до правил належної клінічної практики, затверджених уповноваженим федеральним органом виконавчої влади відповідно до таких цілей:
1) встановлення безпеки лікарських засобів для здорових добровольців та (або) переносимості їх здоровими добровольцями, за винятком таких досліджень лікарських препаратів, вироблених за межами Російської Федерації;
3) встановлення безпеки лікарського препарату та його ефективності для пацієнтів із певним захворюванням, профілактичної ефективності імунобіологічних лікарських препаратів для здорових добровольців;
4) вивчення можливості розширення показань для медичного застосування та виявлення раніше невідомих побічних дій зареєстрованих лікарських препаратів.
2. Щодо відтворених лікарських засобів для медичного застосування проводяться дослідження біоеквівалентності та (або) терапевтичної еквівалентності в порядку, встановленому уповноваженим федеральним органом виконавчої влади.
3. Організацію проведення клінічних досліджень лікарського препарату для медичного застосування вправі здійснювати:
1) розробник лікарського препарату або уповноважена ним особа;
2) освітні організації вищої освіти; організації додаткової професійної освіти;
(Див. текст у попередній редакції)
3) науково-дослідні організації.
4. Клінічні дослідження лікарського препарату для медичного застосування проводяться на підставі дозволу на проведення клінічного дослідження лікарського препарату, виданого уповноваженим федеральним органом виконавчої влади. Уповноважений федеральний орган виконавчої веде реєстр виданих дозволів на проведення клінічних досліджень лікарського препарату, що містить вказівку на їхню мету або цілі, у встановленому цим органом порядку.
(Див. текст у попередній редакції)
(Див. текст у попередній редакції)
6. До організації проведення клінічних досліджень лікарського препарату для медичного застосування розробником лікарського препарату можуть залучатися юридичних осіб будь-якої організаційно-правової форми за умови забезпечення відповідності цих досліджень вимогам цього Закону.
7. Клінічні дослідження лікарських засобів для медичного застосування проводяться в медичних організаціях, акредитованих уповноваженим федеральним органом виконавчої влади в порядку, встановленому Урядом Російської Федерації.
8. Перелік медичних організацій, які мають право проводити клінічні дослідження лікарських препаратів для медичного застосування, та реєстр виданих дозволів на проведення клінічних досліджень лікарських препаратів опубліковуються та розміщуються уповноваженим федеральним органом виконавчої влади в установленому ним порядку на своєму офіційному сайті у мережі "Інтернет".

У 2018 році Різдвяний пост розпочнеться 28 листопада. У цей період православні віруючі готуються до зустрічі Різдва.

Створення сім'ї – мрія більшості жінок. Вони хочуть мати люблячого чоловіка і купу дітей. Тільки от не завжди стосунки...

Ця стаття містить: найсильніша молитва від розлучення - інформація взята зі всіх куточків світла, електронної мережі та...

Інформаційний сайт про ікони, молитви, православні традиції. Молитва від скандалів та сварок у сім'ї, з чоловіком, з дітьми...

Який же Новий рік без шампанського, мандарин, «Олів'є», заливного та всіма улюбленої «Оселедця під шубою». Ось із останнім...

Підготуємо необхідні інгредієнти для печива. Перше, що потрібно зробити - це поставити воду кип'ятитися. Нам необхідний...

Чи можна оформити працівника посаду фінансовий директор - головний бухгалтер. Головний бухгалтер стверджує, що...

Керівник невеликого бізнесу може вести бюджет самостійно. ПЕРЕВІРЕНО! Якщо займатися веденням...

Створення нових проектів передбачає попереднє економічне обґрунтування їхньої доцільності, наступне...
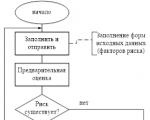
Звітність формується РМ, проходить узгодження (схвалення) Комітету з ризиків за Правління та передається на...

На краю великого луга, на довгій смарагдовій травинці жила крихітна Божа Корівка. Маленька Божа...

Нині досить поширене звернення людини до зірок. За допомогою гороскопу людина може дізнатися...

Діловий чи приятельський. Якщо ж переслідували незнайомку - значить рівень вашої довіри до жіночої статі.

по соннику ФрейдаЯкщо вам снилося, як ви ловили рибу, значить, у реальному житті ви насилу можете відключитися...

Створення сім'ї – мрія більшості жінок. Вони хочуть мати люблячого чоловіка і купу дітей. Тільки от не завжди...

Ця стаття містить: найсильніша молитва від розлучення - інформація взята зі всіх куточків світла, електронної...