Какво може и какво не може по време на Рождественския пост?
През 2018 г. Рождественският пост ще започне на 28 ноември. През този период православните вярващи се подготвят да празнуват Коледа...
Клиничната картина на заболяването е описана за първи път от Андерсен през 1956 г. Заболяването се унаследява по автозомно-рецесивен начин. При тип IV гликогеноза има дефект в ензима амило-1,4 → 1,6-трансглюкозидаза, който участва в образуването на точки на разклонение в молекулата на гликогена:
При тип IV гликогеноза в засегнатите органи се синтезира анормален гликоген, подобен на амилопектина (компонент на нишестето в растителните клетки). Анормалната молекула на гликогена има намален брой точки на разклонение и по-дълги външни и вътрешни вериги в сравнение с нормата.
Заболяването е рядко и е генерализирано (най-често се засягат сърдечната и скелетната мускулатура и черния дроб). Клинично заболяването се проявява с хепатоспленомегалия, асцит, умственото развитие не е засегнато. Прогресивната портална фиброза на черния дроб води до цироза. Цирозата вероятно се развива в резултат на натрупване на амилоподобен гликоген.
Смърт в детска възраст от чернодробна недостатъчност. Патологичното изследване разкрива увеличение на размера на бъбреците, черния дроб и далака. Хепатоцитите са увеличени по размер и съдържат амилопектиноподобен полизахарид.
Лафорова болест- церебрална гликогеноза (миоклонична епилепсия). При това заболяване се открива натрупване на анормален гликоген в мозъка, наподобяващ свойствата на полимера при гликогеноза тип IV. Активността на разклонения ензим не се променя при това заболяване.
Описан за първи път от B. McArdle през 1951 г. Автозомно рецесивен тип наследяване. Характеризира се с дефицит на мускулна фосфорилаза в скелетните мускули. Липсата на мускулна фосфорилаза не се комбинира с нарушение на чернодробната фосфорилаза (контролирана от различни гени). Активността на фосфорилазата на левкоцитите, еритроцитите и тромбоцитите при болестта на McArdle не се променя.
При това заболяване до 3-4% от гликогена, който е нормален по структура, се натрупва в мускулните влакна. Излишният гликоген се отлага под сарколемата в цитоплазмата. В покой, енергийните нужди се осигуряват от миоцитната глюкоза. По време на мускулна работа нуждата от енергийно снабдяване не се задоволява поради ензимен дефект, който причинява болка и спазми при този вид гликогеноза.
Болестта на McArdle е хетерогенна. Клиничните признаци се появяват по-често при възрастни, в детството симптомите на заболяването не са ясно изразени. Заболяването протича в 3 етапа:
1. В детска и юношеска възраст се наблюдава мускулна слабост, умора и възможна миоглобинурия.
2. Между 20 и 40 години мускулните болки стават по-интензивни, а след физическа активност се появяват спазми.
3. След 40 години се появява прогресивна слабост поради мускулна дистрофия.
Установено е, че активността на фосфорилазата рязко намалява при дефицит на витамин В6 (60% от пиридоксина в скелетните мускули е свързан с фосфорилаза). Следователно дефицитът на фосфорилаза влияе върху съдържанието на пиридоксин в организма. Прогнозата за гликогеноза тип V е благоприятна.
Рядко наследствено заболяване от групата на мултисистемните каналопатии. Начинът на унаследяване е автозомно-доминантен, с непълна транспрезия и значителна вариабилност между членовете на едно и също семейство. Спорадичните случаи са чести. Дефектният ген (KCNJ2) се намира на дългото рамо на хромозома 17 (локус 17q23.1-q24.2). Генният продукт участва в образуването на калиеви канали, през които калият навлиза в мускулните клетки. Когато възникне генна мутация, структурата на калиеви канали се нарушава, както и регулацията на навлизането на калиеви йони в клетката (регулаторната молекула PIP2 не може да се свърже с канала). Нарушеното проникване на калиеви йони в мускулните клетки води до развитие на характерни признаци на синдрома (ролята на гена KCNJ2 във формирането на скелетната система все още се проучва). Клинично синдромът се представя от триада от признаци:
характерен дисморфизъм на лицето и скелета;
калий-чувствителна периодична парализа;
камерни аритмии.
Възможно е също увреждане на клапния апарат на сърцето и бъбречна хипоплазия.
Диспластичните характеристики се изразяват в нисък ръст, ниско поставени уши, хипертелоризъм, дефекти на мекото и твърдото небце, хипоплазия на долната челюст, клинодактилия и сколиоза.
Лицето на пациент със синдром на Andersen-Tawil. Заслужават внимание характерните диспластични признаци: хипертелоризъм, хипоплазия на долната челюст и ниско поставени уши (източник Katz J.S., Wolfe G.I., Iannaccone S., Bryan W.W., Barohn R.J. Тестът с упражнения при синдрома на Andersen // Arch. Neurol., 1999. – Vol.56. – P.352-356)
Калий-чувствителната периодична парализа без миотонични прояви, характерни за този синдром, е клинично неразличима от другите форми на хиперкалиемична периодична парализа. Въпреки това, има мнение, че поради изключителната променливост на промените в концентрациите на калий по време на паралитични атаки, традиционните критерии за хипо-, нормо- и хиперкалиемични форми при синдрома на Andersen-Tawil са неприемливи. Често атаките се развиват на фона на продължителна обща слабост.
Сърдечните симптоми включват удължаване на QT интервала с различна тежест, камерна бигеминия, пароксизмална камерна (до бивентрикуларна) тахикардия, внезапен сърдечен арест.
В литературата има съобщения за синдром на внезапна смърт при пациенти, страдащи от това заболяване.
Пациентите често изпитват парадоксални реакции към прилагането на различни лекарства и рефрактерност към антиаритмични средства. Показан е траен положителен ефект от терапията с амиодарон и ацетазоламид (Diacarb) (облекчаване на сърдечни и мускулни симптоми).
За първи път комбинацията от периодична парализа и аритмия е забелязана от Klein et al. през 1963г ( Klein R., Ganelin R., Marks J.F., Usher P., Richards C. Периодична парализа със сърдечна аритмия // J. Pediatr., 1963. – Vol.62. – С.371-385) и Lisak et al. през 1970 г ( Lisak R.P., Lebeau J., Tucker S.H., Rowland L.P. Хиперкалиемична периодична парализа със сърдечна аритмия // Неврология, 1970. – Vol.20. – С.386). Синдромът е описан за първи път от датския лекар Ellen Damgaard Andersen и съавтори през 1971 г. ( Андерсен д. д., Красилников П. А., Overvad з. Прекъсващ мускулест слабост, екстрасистоли и многократни развитие аномалии: а нов синдром? // Acta педиатрица Скандинавия, Стокхолм, 1971. – Vol.60. – П.559–564 ); тя описва случай на заболяването при 8-годишно дете с характерна триада от периодична парализа, аритмия и аномалии в развитието. Впоследствие такава триада е описана само в една работа през 1985 г. И само подробно описание от американски невролог от ливански произход Раби Тауил и др. ( Тавил Р., Птачек Л. Дж., Павлакис С. Ж., DeVivo д. ° С., Пен А. С., Оздемир ° С., Григс Р. ° С. Андерсен‘ с синдром: калий— чувствителен периодичен парализа, вентрикуларен ектопия, и дисморфичен Характеристика // Анали на Неврология, 1994. – Vol.35. – н.3. – П.326-330 ), привлече вниманието на специалистите към тази нозологична форма, стимулирайки нейното по-нататъшно изследване.
13. Priori S. G., Napolitano C., Grillo M. Скрити аритмогенни синдроми: Скритият субстрат на идиопатичната вентрикуларна фибрилация? // Cardiovasc. Рез. - 2001. - кн. 50.
14. Priori S.G., Napolitano C., Memmi M. Клинична и молекулярна характеристика на пациенти с катехоламинергична полиморфна камерна тахикардия // Circulation. - 2002 г.
Vol. 106. - С. 69-74.
15. Priori S.G., Napolitano C., Tiso N. et al. Мутации в гена на сърдечния рианодинов рецептор (hRyR2) са в основата на катехоламинергичната полиморфна камерна тахикардия // Пак там.
2000. - кн. 103. - С. 196-200.
16. Спърджън Д. Внезапните сърдечни смъртни случаи нарастват с 10% при младите американци // Brit. Med. J. - 2001. - Кн. 322. - P. 573 (Резюме).
17. Сумитомо Н., Харада К., Нагашима М. и др. Катехоламинергична полиморфна камерна тахикардия:
© С. М. КРУПЯНКО, Т. Т. КАКУЧАЯ, 2005 УДК 616.12-008.6
СИНДРОМ НА АНДЕРСЕН
С. М. Крупянко, Т. Т. Какучая
Научен център по сърдечно-съдова хирургия на името на. А.
RAMS, Москва
Синдромът на Андерсен е рядка наследствена патология, характеризираща се с преходна мускулна парализа, удължаване на QT интервала, често съчетано с поява на U вълни с висока амплитуда, камерни аритмии и признаци на дисморфогенеза - ниско поставени уши, микрогнатия (ненормално малки челюсти, особено долната челюст), широко чело, клинодактилия -ey (постоянна деформация на един или повече пръсти), синдактилия (сливане или наличие на ремъци между пръстите на краката или ръцете), хипертелоризъм (увеличено разстояние между два чифтни органа), нисък ръст, сколиоза и т.н. През 1971 г. Е. Андерсен и др. съобщава, че 8-годишен пациент има нисък ръст, хипертелоризъм (широко раздалечени очи), хипоплазия на челюстта, широка основа на носа, цепнато небце, скафоцефален череп (дълъг, тесен череп с ръб по протежение на осифицирания сагитален шев), и клинодактилия на петия пръст. През 1994 г. R. Tawil и др. за първи път използва термина „синдром на Андерсен“, за да опише клиничен случай с три характерни черти (клинична триада): чувствителна към калий циклична парализа, нарушения на вентрикуларния ритъм и признаци на дисморфогенеза, наблюдавани от Андерсен през 1971 г. В литературата също често се среща определението „синдром на Андерсен-Тавил“ (Andersen-Tawil syn-
Електрокардиографски характеристики и оптимални терапевтични стратегии за предотвратяване на внезапна смърт // Сърце. - 2003 г.
Vol. 89, № 1. - С. 66-70.
18. Swan H, Laitinen PJ, Kontula K. et al. Антагонизмът на калциевите канали намалява индуцираните от упражнения камерни аритмии при пациенти с катехоламинергична полиморфна камерна тахикардия с RYR2 мутации // J. Cardiovasc. Електрофизиол. - 2005. - кн. 16, № 2. - С. 162-166 (Резюме).
19. Swan H., Piippo K., Viitasalo M. et al. Аритмично разстройство, картографирано към хромозома 1q42-q43, причинява злокачествена полиморфна камерна тахикардия в структурно нормални сърца // J. Amer. Coll. Кардиол. - 1999. - кн. 34, № 7.
20. Тан Х. Л., Хофман Н., Ван Ланген И. М. и др. Внезапна необяснима смърт. Наследственост и диагностична ефективност на кардиологични и генетични изследвания при оцелели роднини // Circulation. - 2005. - кн. 112. - С. 207-213.
Н. Бакулева (директор - академик на Руската академия на медицинските науки Л. А. Бокерия)
дром, съкратено като ATS). Този синдром не трябва да се бърка с болестта на Андерсен, която принадлежи към гликогенозите - заболявания на съхранението на гликоген (поради дефицит на гликоген-конвертиращ ензим, патологично количество гликоген се натрупва в черния дроб, мускулите и други тъкани). Синдромът на Андерсен е първата патология, описана в раздела за каналопатиите - патологии на йонните канали. Синдромът на Андерсен се унаследява по автозомно-доминантен начин, въпреки че има съобщения за спорадични случаи. Вероятността за предаване по наследство е повече от 50%. Тежестта на заболяването може да варира в рамките на едно семейство - едно дете може да има тежки увреждания, докато друго може да бъде клинично асимптоматично. Пенетрантността на заболяването е широко променлива и не всички пациенти проявяват пълния набор от клинични характеристики на този синдром. Нарушенията на ритъма придружават всяка атака на мускулна парализа, възникваща вторично и причинена от резки колебания в нивото на калий в кръвния серум на пациента (фиг. 1). В литературата обаче са описани случаи, при които ритъмните нарушения са били първата клинична проява на синдрома на Андерсен и са предшествали епизоди на мускулна пареза и парализа. При този фенотип на заболяването по-късно е идентифициран удължен QT интервал. Случаи на внезапна сърдечна смърт също са докладвани при синдром на Андерсен.
Анали на аритмологията, № 4, 2005 г
Анали на аритмологията, № 4, 2005 г
III AVF V3 * V6 Серум К+=2,9
III AVF V3 V6 Серум K+=3.4
Ориз. 1. Епизод на вентрикуларна бигеминия при 16-годишно момиче със синдром на Андерсен на фона на хипокалиемия (а) (звездичките показват камерни екстрасистоли); b - нормализирането на нивото на калий в кръвта доведе до изчезването на аритмията.
Серумен К+ - серумен калий.
Въпреки ниското си разпространение в популацията, синдромът на Андерсен е от голям научен и практически интерес за съвременната медицина, тъй като е единственият сред всички генетично определени каналопатии, който засяга както набраздените (скелетни) мускули, така и сърдечния мускул. Други заболявания, които се проявяват като мускулни парези и парализи, се причиняват от мутации в гени, отговорни за транспорта на натриеви, калциеви и калиеви йони в клетките на скелетните мускули, докато различни форми на синдром на удължен QT интервал се причиняват от мутации в гени, кодиращи транспорта на натрий и калиеви йони само в кардиомиоцитите. Предишни проучвания изключват възможността за алелен генезис на синдрома на Андерсен. Въпреки това през 2001 г. N. Plaster et al. При провеждане на молекулярно-генетично изследване в голямо семейство (15 индивида) те откриха връзка между вероятността за наследяване на синдрома на Андерсен и патологията на 17q локуса на хромозома 23 и идентифицираха хетерозиготна миссенс мутация на гена KCNJ2 (17q локуса на хромозома 23 се пресича с локуса на гена KCNJ2 - 17q23.1-17q24 .2) . Мутации на гена KCNJ2 са открити при повече от 50% от пациентите със синдром на Андерсен, което потвърждава факта, че генът KCNJ2 е отговорен за развитието на тази патология. Понастоящем са идентифицирани повече от 20 хетерозиготни миссенс мутации на гена KCNJ2, които причиняват синдром на Andersen (фиг. 2). Гените от семейството KCNJ са широко експресирани в различни тъкани: мускул (KCNJ2, KCNJ11), сърце (KCNJ2, KCNJ3, KCNJ5, KCNJ11), мозък (KCNJ3, KCNJ6, KCNJ9, KCNJ11), епител (KCNJ1, KCNJ2) и много други. Мутациите в генното семейство KCNJ могат да доведат до развитието на три наследствени заболявания при хора и едно заболяване при мишки. Така мутациите на гена KCNJ1 (Kir1.1) водят до развитието на синдром на Bart-ter, автозомно рецесивно заболяване, характеризиращо се с хипокалиемия и загуба на натрий в човешкото тяло; мутациите на гена KCNJ11 (Kir6.2) и свързания протеин SUR1 водят до развитие на постоянна хиперинсулинемична хипо-
Ориз. 2. Структура на субединицата на канала Kir2.1.
Пора - централен отвор (време); М1, М2 - трансмембранни домени; извънклетъчен - извънклетъчен; вътреклетъчен - вътреклетъчен.
Сивите полета показват 20-те мутации, идентифицирани в момента при пациенти със синдром на Андерсен; белият квадрат показва мутацията, идентифицирана в синдрома на късия QT интервал.
гликемия при деца, мутации на Kir3.2 (GIRK2) - до автозомно-рецесивна патология при мишки, проявяваща се със загуба на неврони и тежка атаксия, и накрая, мутациите на гена KCNJ2 са свързани с развитието на синдрома на Андерсен. KCNJ2 кодира синтеза на протеин Kir2.1, който е част от калиевия канал в клетки, способни на възбудимост, включително кардиомиоцити, клетки на набраздения мускул и мозък. Kir2.1 каналите са най-важните регулатори на потенциала на мембраната в покой на сърдечните и скелетните мускули и по този начин възбудимостта на клетките в тези тъкани като цяло, тъй като те осигуряват освобождаването на калиеви йони от клетки с хиперполяризирана мембрана по време на крайния фаза на реполяризация на акционния потенциал. Протеинът Kir2.1 се състои от 427 аминокиселини с два трансмембранни домена (M1, M2) и централна дупка (пора) (H5) и регулира IK1 компонента на калиевия ток със забавена ректификация на фазата на реполяризация (виж Фиг. 2 ). Анализът на Northern blot разкри 5,5-kb транскрипт на гена KCNJ2 с високи нива на Kir2.1 в сърцето, мозъка, плацентата, белите дробове и скелетните мускули и по-ниски нива в бъбреците. В сърцето Kir2.1 каналите преобладават в предсърдията, вентрикулите (с висока скорост на провеждане на тока IK1) и влакната на Purkinje и са по-рядко срещани в нодалните клетки. Kir2.1 субединиците, кодирани от гена KCNJ2, се комбинират в тетрамер в клетъчната стена на кардиомиоцитите, образувайки функциониращ канал; те могат също да се комбинират с други субединици от семейството Kir2.1 като хетеромултимери, което показва тяхната функционална сложност и разнообразие. Повечето KCNJ2 генни мутации са миссенс мутации.
с доминантно-отрицателен ефект, което води до намаляване на тока IK1, намаляване на реполяризацията и увеличаване на продължителността на потенциала на действие. В резултат на горните процеси настъпва деполяризация и дестабилизация на мембранния потенциал на покой, което инициира появата на камерни аритмии или миотонични контракции на скелетните мускули. Освен това изследователите предполагат, че дисфункцията на канала Kir2.1 и намаляването на тока IK1 играят важна роля в развитието на сигнални дисфункции в невъзбудими тъкани, което може да обясни проявите на дисморфогенеза при пациенти със синдром на Андерсен. M. Tristani-Firouzi и др. изследва функционалните последици от мутациите в синдрома на Андерсен чрез експресиране на променения протеин in vitro. Наблюдавано е значително увреждане на функцията на канала Kir2.1 при всички видове мутации, идентифицирани до момента.
Клиничните прояви на синдрома на Андерсен са изключително променливи, което се обяснява с наличието на различни мутации, които допринасят за проявата на различни фенотипове на заболяването. Вероятно дефектите в други йонни канали също са важни в етиологията на синдрома на Андерсен. Повтарящата се мускулна парализа може да бъде причинена от патология на калциевите, натриевите и калиевите канали на миоцитите. Пациентите изпитват краткотрайни епизоди на слабост в ръцете и краката, до генерализирана пареза и парализа на крайниците. Такива атаки могат да се появят в покой, след тренировка или след събуждане сутрин. Трябва да се отбележи, че провокиращите фактори за заболяването са различни. Понастоящем не са известни всички хранителни фактори, които провокират атака на мускулна парализа при синдрома на Андерсен. Задействащите фактори вероятно включват храни, богати на калий и глюкоза. Други фактори, които провокират появата на периодична мускулна парализа, могат да бъдат: промяна в дейностите - почивка след физическа активност, физическа активност след дълъг период на седене, събуждане след сън, дълга разходка на гладно, обилно хранене. Всяка настинка също може да причини мускулна парализа; В литературата има съобщения за случаи, при които провокаторите са били вдишани газове - въглероден диоксид, бензинови пари или дори миризмата на блажна боя. В проучването Tristani-Firuozi, циклична мускулна парализа е наблюдавана при 23 (64%) от 36 пациенти с KCNJ2 генни мутации. Почивката след физическа активност е най-честият тригер за възникване на мускулна парализа, както при класическите варианти от този тип.
състояния, като по-голямата част (55%) от пациентите имат хипокалиемия (серумна концентрация на калий по-малка или равна на 3,4 mEq/L), хиперкалиемия се наблюдава при 22%, а нормокалиемия при 10% от пациентите. Въпреки това, при синдрома на Андерсен хипокалиемичните състояния не са предшествани от приема на въглехидрати, какъвто е случаят с класическите варианти на хипокалиемична мускулна парализа. Мускулна биопсия, извършена при 12 пациенти, разкри неспецифични промени - леки миопатии и/или тубулни агрегати, наблюдавани при други форми на циклична мускулна пареза или парализа. Инхибиторите на въглеродната анхидраза са ефективни при намаляване на честотата на пристъпите на парализа при синдрома на Andersen, както и при класическите форми на мускулна парализа.
Най-характерните промени в ЕКГ при пациенти със синдром на Andersen са удължаване на OT интервала и високоамплитудни вълни и (фиг. 3). Понастоящем има противоречиви мнения сред изследователите относно участието на синдрома на Андерсен в един от вариантите на синдрома на дългия OT интервал. Тъй като синдромът на Andersen се счита за генетично обусловена патология на реполяризацията на мускулните клетки, той започва да се класифицира като синдром на дълъг OT интервал (LTS), а именно тип 7 (LOTS). Понастоящем са идентифицирани 5 гена, мутациите в които са надеждно отговорни за развитието на типични клинични прояви на синдрома на дългия OT интервал: KSYO1 (SHT1), IEAO (KSYN2, SHT2), SSY5L (SHT3), KSYO1 (SHT5), KSYO2 (SHT6). Подобно на други форми на вроден синдром на удължен OT, първичната проява е странична
Ориз. 3. Най-типичните електрокардиографски промени и ритъмни нарушения, открити при пациенти със синдром на Андерсен. а - удължаване на OT интервала; б - краткотрайна нестабилна полиморфна камерна тахикардия, последвана от камерна бигеминия; в - двупосочна камерна тахикардия; d - стрелките показват изпъкнали зъби и.
Анали на аритмологията, № 4, 2005 г
Анали на аритмологията, № 4, 2005 г
Основната характеристика на сърдечно-съдовата система при синдрома на Андерсен е удължаването на OT интервала, идентифицирано при 71% от всички носители на гена KSH2. Средната стойност на коригирания OT интервал (OTI) при мъжки и женски пробанд със синдром на Andersen е съответно 479 и 493 ms, докато при други форми на дълъг OT синдром е съответно 497 и 510 ms. Поради наличието на удължен OT интервал и възможността за развитие на животозастрашаващи камерни аритмии, някои изследователи смятат синдрома на Andersen за един от подтиповете на вроден удължен OT синдром. Въпреки това, както се оказа, въпреки честата поява на вентрикуларни аритмии (вентрикуларни екстрасистоли и серии от непродължителни камерни тахикардии, включително двупосочни камерни тахикардии, вижте фиг. 3) - при 64% от пациентите в проучването M. Il81ash-R1goi21, рискът от внезапна сърдечна смъртност при синдрома на Andersen е по-нисък, отколкото при синдрома на дългия OT интервал и други наследствени каналопатии. За да идентифицират механизмите на възникване на вентрикуларни аритмии при синдрома на Андерсен, авторите симулираха потискането на функцията на канала Yu2.1 на кардиомиоцитите на вентрикула на сърцето в експеримент. Оказа се, че потискането на функцията на канала Yu2.1 на фона на хипокалиемия води до удължаване на крайната фаза на акционния потенциал на сърдечния мускул, поява на забавени постдеполяризации поради индуцирането на Ca + / Ca2 + обмен и развитие на спонтанни аритмии. Авторите заключават
че субстратът за появата на вентрикуларни аритмии при синдрома на Andersen се различава от този при други форми на синдром на вроден дълъг OT интервал и е по-близък до аритмиите в резултат на претоварване с Ca2+ или дигиталисова интоксикация (като двупосочна и полиморфна камерна тахикардия). По този начин при синдрома на Андерсен се комбинират клиничните прояви на вроден синдром на дълъг OT интервал и фамилна (катехоламинергична) полиморфна камерна тахикардия (с последния, като правило, не се наблюдава удължен OT интервал). Диапазонът от ритъмни нарушения, открити при синдрома на Андерсен, е представен в таблицата.
Високата амплитуда на вълните е специфична находка при синдрома на Andersen и се регистрира главно в предните гръдни отвеждания (виж Фиг. 3). Тази черта е открита в 76% от пробандите и 47% от носителите на мутантни KSH2 гени според М. ln81at-Igou21. Трябва да се отбележи, че обикновено при здрави хора вълната и може да се регистрира при рядка сърдечна честота, докато при пробандите със синдром на Андерсен в изследването на M. ln81at-Igoi21 средната сърдечна честота е 84±17 удара/мин (от 52 до 115 удара/мин).min). Известно е, че при хипокалиемия амплитудата на вълната също може да бъде висока, но не можахме да намерим никакви проучвания, отчитащи нивото на калий в кръвния серум при пациенти с вълни с висока амплитуда, така че ролята на хипокалиемията в генезиса на вълни с висока амплитуда и при синдром на Андерсен не могат да бъдат изключени. Автоматичен-
Клинични прояви на синдрома на Андерсен при пробанди с мутантен KSP ген
Мутационен клъстер Пол Сърдечна честота Продължителност OT, ms Ритъмни нарушения
D71V 4415 F 100 513 Не е открит
A95-98 (A-делеция) 3328 F 83 475 Бигеминия, полиморфен VT
8136B 6634 F 68 500 Бигеминия
P186b 7246 M 94 PBPNPG* Полиморфен VT
R218W 2401 F 100 560 Бигеминия, нефатален сърдечен арест, AV блок 1-ва степен, фибрилация, камерно трептене
R218W 2679 M 68 510 Бигеминия, полиморфен VT
R218W 2681 F 75 488 Bigeminy, полиморфен VT
R218W 7480 M 94 525 Бигеминия
R218W 6515 M 52 416 Бигеминия, полиморфен VT
R218Q 6562 M 70 469 Не е открит
G300V 3677 F 100 480 Бигеминия, полиморфен VT
V302M 2682 M 79 PBPNPG* Бигеминия
E303K 2281 F 85 495 Чести камерни екстрасистоли
A314-315 5768 F 115 471 Фибрилация, камерно трептене, мономорфна VT
* Пълният десен блок на бедрата възпрепятства точното измерване на OT интервала.
Изследователите също така оценяват връзката между тежестта на мутациите (тежестта на дисфункцията на Kr2.1 канала) и тежестта на клиничните прояви (удължаване на TPV, аритмии, невромускулни симптоми, дисморфогенеза) на синдрома на Андерсен. Оказа се, че клиничният фенотип на заболяването не корелира със степента на доминантно-отрицателно потискане на функцията на K1r2.1 канала.
G. Andelfinge и др. идентифицира хетерозиготната миссенсна мутация R67W на гена KSH2 в 41 кръвни роднини и се оказа, че унаследяването на камерни аритмии и периодична мускулна парализа се различава в зависимост от пола: по този начин камерните аритмии преобладават при жените (13 от 16, или 81%) , а невромускулни симптоми - при мъже (10 от 25, или 40%). В същото време вентрикуларните аритмии започват да се появяват след 10-годишна възраст и са по-редки по време на бременност (докато при други видове генни мутации на KSH2 при жените ритъмните нарушения са по-чести по време на бременност) и след 55 години (по време на менопауза). Трябва да се отбележи, че няма удължаване на OT интервала в това родословие. В тази група пациенти трима са имали анамнеза за синкоп; На един пациент, оцелял след внезапна сърдечна смърт, е имплантиран кардиовертер-дефибрилатор. При 8 пациенти от мъжки пол са наблюдавани епизоди на мускулна слабост или парализа след физическо натоварване, а при 2 е имало хипокалиемия. Хипертелоризъм се наблюдава при 4 индивида, малка долна челюст при 10, синдактилия на пръстите на ръцете или краката при 9, клинодактилия при 12. Признаците на дисморфогенеза са еднакво чести при мъже и жени. Един пациент е опериран за сколиоза и една пациентка за цепнато небце. Много необичайно беше идентифицирането в изследваното родословие на едностранна бъбречна дисплазия и клапна патология на сърцето - стеноза на белодробна клапа (диагностицирана при един пациент на 6-месечна възраст), бикуспидна аортна клапа (при 3 пациенти) и бикуспидна аортна клапа с коарктация на аортата (при 1 пациент). Такива аномалии са идентифицирани за първи път при пациенти със синдром на Андерсен. Освен това има съобщение за смърт на новородено с неизвестен сърдечен порок. Атриовентрикуларен (AV) блок първа степен е наблюдаван при 4 мъже и една жена в комбинация с левия бедрен блок. Нито един човек не е имал едновременно всички прояви, характерни за синдрома на Андерсен. Плейотропия на фенотипните прояви при синдрома на Андерсен в рамките
Това родословие (фиг. 4) може да се обясни или със специфичния ефект на мутацията R67W, или с вариация в експресията на алелите, или с модифициращия ефект на външни фактори.
Има няколко описания на лечения, използвани за синдрома на Андерсен. Като пример цитираме доклада на J. Junker et al. : при 6-годишен пациент без фамилна анамнеза за наследствени или сърдечно-съдови заболявания за първи път започнаха да се появяват повтарящи се епизоди на атонична пареза. На 10-годишна възраст тя е подозирана за постстрептококов миокардит поради високо ниво на серумна креатиназа (до 447 U/L) и асимптоматични полиморфни камерни екстрасистоли (PVCs), записани на ЕКГ. Поради спонтанно спиране на пристъпи на мускулна слабост, лекарите не изключват възможността за техния психогенен произход. На 15-годишна възраст пациентът е преживял епизод на камерно мъждене (VF) и успешно е извършена кардиопулмонална реанимация. Програмираната електрическа стимулация предизвиква продължителна камерна тахикардия с QTc от 0,45 s. На пациента е имплантиран кардиовертер-дефибрилатор (ICD). След една година започнаха да се появяват ежедневни пристъпи на VF, изискващи ICD шокове, и бяха наблюдавани епизоди на тежка мускулна слабост (дори мускулна парализа). Сред характеристиките, които привлякоха вниманието по време на физическия преглед, бяха: широка основа на носа, клинодактилия, сколиоза и нисък ръст. Атоничната мускулна парализа обхваща различни мускулни групи на горните и долните крайници, възниква внезапно, продължава няколко часа или дни и спира от само себе си. Нивата на серумния калий и креатинкиназата са нормални. Пристъпите на слабост са предизвикани от хиперкалиемия или настинка, а упражненията са придружени от намаляване на мускулния потенциал за действие. Мускулната биопсия разкрива тубуларни агрегати и няколко вакуоли. Клиничната диагноза на спорадичния вариант на синдрома на Андерсен беше потвърдена след молекулярно-генетично изследване, което разкри хетерозиготна миссенсна мутация R218W на гена KCNJ2 при момичето, но не и при родителите му. Соталол и антиаритмичните лекарства от клас I (ААА), включително флекаинид и пропафенон, са неефективни. Към терапията с каптоприл, надолол и дигитоксин беше добавен амиодарон в натоварваща доза от 200 mg дневно. Вентрикуларните аритмии бързо изчезнаха и пристъпите на мускулна слабост продължиха да се повтарят. След 2 месеца към горната терапия беше добавен ацетазоламид (Diamox) в доза 750 mg на ден. През следващите 2 години пациентът
Анали на аритмологията, № 4, 2005 г
Анали на аритмологията, № 4, 2005 г
1 съобщават или за неуспех на терапията с амиодарон и ацетазоламид при пациенти със синдром на Andersen, или за необходимостта от преустановяване поради развитието на странични ефекти. От друга страна, може да има фармакогенетично взаимодействие между мутацията R218W и терапевтичния отговор към амиодарон и ацетазоламид, което изисква потвърждение при други пациенти. Тъй като амиодаронът инхибира IK1 тока, логично е да се предположи, че той инхибира свръхвъзбудимостта на сърдечния мускул чрез забавяне на функцията на натриевите и калциевите канали, както и на бета-адренергичните рецептори.
канавка, като по този начин коригира промените, причинени от загубата на функция на канала South2.1. Въпреки това, поради потенциала за значителни странични ефекти, амиодарон може да се препоръча при пациенти със симптоматични аритмии. Ацетазоламид, лекарство, което променя киселинността на кръвта, може да предотврати пристъпи на периодична мускулна парализа от всякакъв произход поради селективното отваряне на мускулните Ksa2+ канали, което може да компенсира дисфункцията на тока IK1 и да предотврати спадане на мембранния потенциал на мускулите при синдром на Андерсен. Пациенти с хипокалиемични форми на мускулна пареза могат да приемат калиев хлорид, разтворен в неподсладен разтвор (симптомите обикновено изчезват в рамките на един час); те трябва да избягват храни, богати на въглехидрати и прекомерни упражнения. Пациентите с хиперкалиемични форми на мускулна парализа могат да предотвратят тези атаки, като ядат чести храни, богати на въглехидрати и бедни на калий.
По този начин ние представихме най-изчерпателния преглед досега на клиничната картина, диагнозата и лечението на синдрома на Андерсен, една от уникалните патологии сред наследствените каналопатии поради необичайната комбинация от фенотипни прояви на сърдечната и мускулно-скелетната система в комбинация с различни
фигуративни признаци на дисморфогенеза, чийто механизъм на възникване все още остава неясен. Подобно на откриването на нови варианти на такива генетично обусловени заболявания като синдром на дълъг или къс QT интервал, по-нататъшни изследвания в областта на молекулярната генетика ще помогнат за идентифицирането на нови форми на синдрома на Андерсен и ще проучат по-задълбочено механизмите на действие на мутациите (мутациите могат да нарушат функции на йонни канали и трансмембранни протеини на различни нива на тяхното взаимодействие) и обясняват необичайните фенотипни прояви на това заболяване, а фармакогенетичните изследвания ще помогнат за разработването на нови методи за лечение.
ЛИТЕРАТУРА
1. Andersen E.D., Krasilnikov P.A., Overad H. Периодична мускулна слабост, екстрасистоли и множество аномалии в развитието: Нов синдром? // Acta Pediatr. Сканиране.
1971. - кн. 60. - С. 559-564.
2. Andelfinger G., Tapper A. R., Welch R. C. et al. KCNJ2 мутацията води до синдром на Андерсен със специфични за пола сърдечни и скелетни мускулни фенотипове // Amer. J.Hum. Женет. - 2002. - кн. 71. - С. 663-668.
3. BakerN., Iannaccone S.T., BurnsD., Scott W. Andersen’s syn-
дром: Епизодична слабост и фамилна вентрикуларна дисритмия // J. Child. неврол. - 1996. - Вл. 11. - С. 152 (Абстр.).
4. Bendahhou S., Donaldson M. R., Plaster N. M. et al. Дефект-
тивният трафик на калиев канал Kir2.1 е в основата на синдрома на Andersen-Tawil // J. Biol. Химия. - 2003. - кн. 278, № 51. - P. 51779-51785.
5. Bosch R. F., Li G. R., Gaspo R., Nattel S. Електрофизиологични ефекти на хроничната терапия с амиодарон и хипотиреоидизъм, самостоятелно и в комбинация, върху вентрикуларни миоцити на морско свинче // J. Pharmacol. Exp. Там. - 1999. - кн. 289. - С. 156-165.
6. Canun S., PerezN., Beirana L.G. Автозомно доминантен синдром на Andersen в три поколения // Amer. J. Med. Женет. - 1999. - кн. 85. - С. 147-156.
7. Jen J., Ptacek L. J. Channelopathies // Метаболитни и молекулярни основи на наследствено заболяване / C. R. Scriver, A. L. Beaudet, W. S. Sly, D. Valle (eds). - N.Y.: McGraw-Hill, 2001.- P. 5223-5238.
ne и ацетазоламид за лечение на генетично потвърден тежък синдром на Андерсен // Неврология. - 2002 г.
Vol. 59, № 3. - С. 466.
9. Lange P. S., Er F., Gassanov N., Hoppe U. C. Andersen
мутациите на KCNJ2 потискат нативния вътрешен токоизправител
ток IK1 по доминантно-отрицателен начин // Cardiovasc. Рез. - 2003. - кн. 59, № 2. - С. 321-327.
10. Kimbrough J. et al. Клинични последици за засегнатите родители и братя и сестри на пробанди със синдром на дълъг QT // Circulation. - 2001. - кн. 104. - С. 557-562.
11. Klein R, Genelin R., Marks J. F. Периодична парализа със сърдечна аритмия // J. Pediatr. - 1965. - кн. 62. - С. 371-385.
12. Keating M. T., Sanguinetti M. C. Молекулярни и клетъчни механизми на сърдечни аритмии // Cell. - 2001. - кн. 104.
13. Левит Л. П., Роуз Л. И., Доусън Д. М. Хипокалиемична периодична парализа с аритмия // N. Engl. J. Med. - 1972 г.
Vol. 286. - С. 253-254.
14. Гипс Н. М. и др. Мутациите в Kir2.1 причиняват развитието и епизодичните електрически фенотипове на синдрома на Андерсен // Cell. - 2001. - кн. 105. - С. 511-519.
15. Pouget J., Philip N., Faugere G., Pellissier J. F. Andersen syndrome: Особена форма на парализа със сърдечна аритмия // Rev. неврол. (Париж). - 2004. - кн. 160, № 5 (Pt. 2). - S. 38-42 (френски).
16. Sansone Ket al. Синдром на Андерсен: отчетлива периодична парализа // Ann. неврол. - 1997. - кн. 42. - С. 305-312.
17. Schulze-Bahr E. Синдром на къс QT интервал или синдром на Andersen: Ин и Ян на дисфункция на канала Kir2.1 // Circ. Рез. - 2005. - кн. 96. - С. 703-704.
18. Surawicz B. U вълна: факти, хипотези, погрешни схващания и погрешни наименования // J. Cardiovasc. Електрофизиол. - 1998 г.
Vol. 9. - С. 1117-1128.
19. Tawil R. et al. Синдром на Андерсен: чувствителна към калий периодична парализа, вентрикуларна ектопия и дисморфични характеристики // Ann. неврол. - 1994. - кн. 35. - С. 326-330.
20. Tawil R. et al. Рандомизирани проучвания на дихлорфенамид при периодични парализи. Работна група по периодична парализа // Пак там. - 2000. - кн. 47. - С. 46-53.
21. Tricarico D., Barbieri M., Conte Camerino D. Acetazolamide отваря мускулния KCa2+ канал: Нов механизъм на действие, който може да обясни терапевтичния ефект на лекарството при хипокалиемична периодична парализа // Пак там. - 2000. - кн. 48.
22. Tristani-Firouzi M., Jensen J.L., Donaldson M.R. et al. Функционална и клинична характеристика на KCNJ2 мутации, свързани с LQT7 (синдром на Андерсен) // J. Clin. Инвестирам. - 2002. - кн. 110, № 3. - С. 381-388.
UDC 616.124.3:616.127]-07-08
ДИАГНОСТИКА, ПРОТИЧАНИЕ И ЛЕЧЕНИЕ НА АВТОЗОМНО-ДОМИНАНТНА АРИТМОГЕННА КАРДИОМИОПАТИЯ/ДИСПЛАЗИЯ НА ДЯСНА КАМЕРА
Л. А. Бокерия, В. А. Базаев, А. Х. Меликулов, У. Т. Кабаев, О. Л. Бокерия, Р. В. Висков,
А Г. Филатов, А Н. Грицай, В. В. Чумаков
Научен център по сърдечно-съдова хирургия на името на. А. Н. Бакулева (директор - академик на Руската академия на медицинските науки Л. А. Бокерия) RAMS, Москва
ритмогенна дисплазия на дясната камера или аритмогенна дясна вентрикуларна кардиомиопатия/дисплазия - патология с неизвестна етиология, често наследствена, характеризираща се с животозастрашаваща
тъпа или фибромастна инфилтрация на миокарда, предимно в панкреаса, придружена от камерни аритмии с различна тежест, включително камерно мъждене.
Анали на аритмологията, № 4, 2005 г
Гликогеноза тип IV (болест на Андерсен, амилопектиноза, дифузна гликогеноза с чернодробна цироза)- наследствено заболяване, което се причинява от дефицит на ензими, участващи в метаболизма на гликогена; характеризиращ се с нарушение на структурата на гликогена, неговото недостатъчно или прекомерно натрупване в различни органи и тъкани.
Болест на Андерсенвъзниква в резултат на мутации в гена на микрозомалната амило-1,4:1,6-глюкан трансфераза, което води до неговия дефицит в черния дроб, мускулите, левкоцитите, еритроцитите и фибробластите. Генът е картографиран върху хромозома 3p 12. Начинът на унаследяване е автозомно рецесивен.
Амило-1,4:1,6-глюкан трансферазата участва в синтеза на гликоген в разклонителните точки на гликогеновото дърво. Ензимът свързва сесия от най-малко шест α-1,4-свързани глюкозидни остатъка на външните гликогенови вериги към гликогеновото „дърво“ чрез α-1,6-гликозидна връзка. Когато ензимът е дефицитен, амилопектинът се отлага в чернодробните и мускулните клетки, което води до увреждане на клетките. Концентрацията на гликоген в черния дроб не надвишава 5%.
болестсе проявява през първата година от живота с неспецифични стомашно-чревни симптоми: повръщане, диария. С прогресирането на заболяването се появяват хепатоспленомегалия, прогресивна чернодробна недостатъчност, генерализирана мускулна хипотония и атрофия и тежка кардиомиопатия. Смъртта на пациентите обикновено настъпва преди 3-5 годишна възраст поради хронична чернодробна недостатъчност, рядко в по-напреднала детска възраст (до 8 години).
Лабораторна диагностикавъз основа на откриване на гликоген с променена структура в чернодробна биопсия и намаляване на активността на амило-1,4:1,6-глюкан трансфераза.
Лечението е насочено към борба с метаболитни нарушения, вкл. с ацидоза. В някои случаи е ефективно използването на глюкагон, анаболни хормони и глюкокортикоиди. При хипогликемия са необходими чести хранения с високо съдържание на лесно смилаеми въглехидрати. При мускулни форми на гликогеноза се отбелязва подобрение чрез спазване на диета с високо съдържание на протеини, прилагане на фруктоза (перорално 50-100 g на ден), мултивитамини и ATP. Правят се опити да се прилагат липсващи ензими на пациентите.
Пациентите с гликогеноза подлежат на диспансерно наблюдение от лекар в медицинския генетичен център и педиатър (терапевт) в клиниката.
Превенцията не е разработена. За да се предотврати раждането на дете с гликогеноза в семейства, където има подобни пациенти, се провежда медицинско и генетично консултиране.
Болестта на Андерсен (гликогеноза тип IV, амилопектиноза) възниква поради дефицит на ензима за разклоняване на 1,4-а-глюкан, което води до натрупване на необичаен, слабо разтворим гликоген.
Това заболяване се нарича амилопектиноза, тъй като гликогенът в такива случаи е по-малко разклонен и има по-дълги линейни участъци, съдържащи α-1,4-гликозидни връзки, което е характерно за структурата на амилопектина.
Болестта на Андерсен се унаследява по автозомно-рецесивен начин. Генът на 1,4-а-глюкан разклонения ензим е разположен на хромозома 3; Неговите мутации в основата на заболяването са известни и техните характеристики във всеки отделен случай позволяват да се предвиди клиничната картина на заболяването.
Симптоми на болестта на Андерсен
Амилопектинозата е клинично хетерогенна. Най-честата му класическа форма се характеризира с прогресираща цироза на черния дроб. Първоначалните признаци - хепатоспленомегалия и слабо развитие - се появяват през първите 18 месеца. живот. Постепенно се развиват портална хипертония, асцит, разширени вени на хранопровода и чернодробна недостатъчност, от които пациентите умират до 5-годишна възраст. В редки случаи увреждането на черния дроб не прогресира.
Има съобщения и за невромускулна форма на болестта на Андерсен. Проявите му са разнообразни:
дифузно увреждане на централната, периферна нервна система, придружено от натрупване на полиглюкозови телца в невроните (така наречената болест на полиглюкозовите телца) Диагнозата в последния случай изисква определяне на активността на 1,4-а-глюкан-разклоненията ензим в левкоцити или биопсии на нервна тъкан, тъй като дефицитът му е ограничен именно в тези клетки.
Диагностика на болестта на Андерсен
Отлагането на атипичен гликоген се открива в черния дроб, сърцето, мускулите, кожата, червата, главния и гръбначния мозък и периферните нерви. В черния дроб се развива малка нодуларна цироза. При изследване се виждат слабо оцветени базофилни включвания в хепатоцитите, които са едрозърнести PAS-положителни отлагания, частично устойчиви на амилаза. Електронната микроскопия разкрива, в допълнение към гликогенните частици, фиброзни агрегати, характерни за амилопектина. Характерното оцветяване на цитоплазмените включвания и електронномикроскопската картина могат да имат диагностична стойност, но подобни хистологични характеристики се наблюдават при полизахаридози без дефицит на ензима за разклоняване на 1,4-а-глюкан. За потвърждаване на диагнозата е необходимо да се установи дефицит на този конкретен ензим в черния дроб, мускулите, култивирани кожни фибробласти или левкоцити. За целите на пренаталната диагностика, активността на разклонения ензим 1,4-a-glucan се определя в култивирани амниоцити или хорионни въси.

През 2018 г. Рождественският пост ще започне на 28 ноември. През този период православните вярващи се подготвят да празнуват Коледа...

Създаването на семейство е мечтата на повечето жени. Те искат да имат любящ съпруг и куп деца. Но не винаги е връзка...

Тази статия съдържа: най-силната молитва за развод - информация, взета от всички краища на света, електронната мрежа и...

Информационен сайт за икони, молитви, православни традиции. Молитва за скандали и кавги в семейството, със съпруга, с децата...

Каква би била Нова година без шампанско, мандарини, оливие, заливка и любимата на всички „Херинга под шуба“. С последното...

Нека подготвим необходимите съставки за бисквитките. Първото нещо, което трябва да направите, е да сложите водата да заври. Имаме нужда от...

Възможно ли е записване на служител за длъжността финансов директор - главен счетоводител? Главният счетоводител твърди...

Ръководителят на малък бизнес може лесно да управлява бюджета самостоятелно. ПРОВЕРЕНО! Ако успееш...

Създаването на нови проекти е свързано с предварителна икономическа обосновка за тяхната осъществимост, последващо...
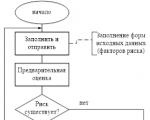
Отчетността се генерира от RM, съгласува се (одобрява) от Комитета по риска към Управителния съвет и се предава на...

В края на голяма, много голяма поляна, върху дълга изумрудена трева живееше мъничка калинка. Боже малко...

В днешно време е доста обичайно хората да се обръщат към звездите. С помощта на хороскоп човек може да разбере...

Бизнес или приятелски. Ако сте преследвали непознат, това означава нивото на доверие в женския пол...

според съновника на Фройд Ако сте сънували как ловите риба, това означава, че в реалния живот трудно можете да изключите...

Създаването на семейство е мечтата на повечето жени. Те искат да имат любящ съпруг и куп деца. Но не винаги...

Тази статия съдържа: най-силната молитва за развод - информация взета от цял свят, електронна...