Kas galima ir ko negalima per Gimimo pasninką?
2018 metais gimimo pasninkas prasidės lapkričio 28 d. Šiuo laikotarpiu stačiatikiai ruošiasi švęsti Kalėdas...
Sveiki vėlgi į projekto „Mamos prieš geležies trūkumą“ puslapius! Tačiau šiandien kalbėsime ne apie jį, o apie ligą, kuri bendrame kraujo tyrime labai panaši į geležies trūkumą. Pakalbėkime apie talasemiją.
Liga, kaip matyti iš apibrėžimo, dažniausiai paveldima iš tėvų. Jis atsiranda dėl klaidos (mutacijos) genuose, kurie koduoja hemoglobino molekulės struktūrą mūsų organizme. Žmonėms, turintiems tokį „sulaužytą“ geną, hemoglobinas gaminasi su pakitusiomis savybėmis. Dėl to raudonieji kraujo kūneliai (raudonieji kraujo kūneliai) atrodo labai maži ir blyški žiūrint pro mikroskopą. Be to, šie raudonieji kraujo kūneliai yra labai lengvai sunaikinami, palyginti su įprastais. Iš šito žmonėms, sergantiems sunkiomis ir vidutinio sunkumo talasemijos formomis, hemoglobino kiekis periodiškai smarkiai sumažėja ir raudonųjų kraujo kūnelių kraujyje ir atsiranda gelta(ši būklė vadinama hemolize – staigiu raudonųjų kraujo kūnelių sunaikinimu). Iš sunaikintų raudonųjų kraujo kūnelių į kraują patenka daug laisvo hemoglobino, kurį kepenys greitai paverčia bilirubinu. O bilirubinas odą ir gleivines, įskaitant akių baltymus, nuspalvina ryškiai geltonai (tai gelta, panašiai kaip ir naujagimiams).
Staigią hemolizę patyrusiam vaikui galite padėti donoro kraujo perpylimu. Bet tik laikinai, iki kitos hemolizės (raudonųjų kraujo kūnelių sunaikinimo), kuri dažnai atsiranda reaguojant į virusinę infekciją ar kitą stresą. O kartais tiesiog be jokios aiškios priežasties.
Sunkiausiais atvejais radikalus talasemijos gydymas gali būti kaulų čiulpų transplantacija.
Bet sunki ar kitokia pagrindinė talasemijos forma laimei, Rusijoje tai reta! Daug dažniau diagnozuoja vaikų hematologai nedidelė talasemijos forma ir ji beveik neturi jokių simptomų daugumai mūsų jaunų pacientų. Todėl pediatrui sunku nustatyti teisingą diagnozę, kaip jau sakiau, iš pradžių mažąją talasemiją galima lengvai supainioti su geležies stokos anemija. Todėl šiandien tam skirsime ypatingą dėmesį.
Kiekviena jūsų ląstelė turi 46 chromosomas, tiksliau – 23 poras chromosomų. Kiekvienoje poroje viena chromosoma atkeliavo pas jus iš mamos, kita - iš tėvo. Kiekvienoje motinoje yra išties išskirtinių ląstelių – tai kiaušialąstės, jose yra ne 46, o tik 23 chromosomos. Tokių išimčių turi ir mūsų vaikų tėčiai – tai spermatozoidai, kurie taip pat turi 23 chromosomas. Kai apvaisinimo momentu spermatozoidų ir kiaušialąstės branduoliai susilieja, kiekviena iš 23 motinos chromosomų randa atitinkamą iš 23 tėvo chromosomų ir sudaro tokias pačias poras. Pasirodo, kiekviena jūsų būsimo kūdikio ląstelė vėl turi 46 chromosomas.
Kodėl viskas taip sudėtinga? Gamta turėjo daug priežasčių, ir viena iš jų buvo mūsų saugumas.
Paprastai kiekvienoje iš suporuotų chromosomų yra tie patys genai. Bet jei vienas iš tėvų netyčia turi paklaidą (mutaciją) vietoj vieno iš šių genų, tada iš antrojo tėvo gautas genas apdraus, ir organizmas galės normaliai funkcionuoti.
Pažvelkime į pavyzdį naudojant tą patį šeimos medį.
 Legenda: apskritimas – moteris, kvadratas – vyras, kryžius – talasemijos genas, nulis – sveiko genas. Rodyklės rodo genų perdavimą iš kartos į kartą.
Legenda: apskritimas – moteris, kvadratas – vyras, kryžius – talasemijos genas, nulis – sveiko genas. Rodyklės rodo genų perdavimą iš kartos į kartą. Moteris (A) yra talasemijos geno nešiotoja, kuri iš tikrųjų niekada neturėjo talasemijos, tačiau oficiali diagnozė bus: talasemija, nedidelė forma. Šį geną ji gavo, pavyzdžiui, iš savo tėčio, kuris taip pat niekada neturėjo simptomų, todėl apie šią ligą šeimoje niekas nežino. Ji turi antrą atitinkamą geną iš mamos, jis yra sveikas.
Moteris ištekėjo už vyro (B), kuris (yra tokių sutapimų!) taip pat turi vieną sergantį geną iš tėvo (C), o sveiką – iš mamos. Jie pastojo ir pagimdė berniuką (D), kuris iš mamos paveldėjo sveiką geną, o iš tėvo – talasemijos geną. Taigi berniukas sirgs tokia pačia lengva ar besimptome ligos forma kaip ir jo tėvai, nors jis yra vieno ligos geno nešiotojas.
Bet tada jie pastojo antrą kartą. Jie turi gimė dar vienas berniukas (E), kuris paveldėjo „sulaužytus“ genus ir iš savo tėvo, ir iš mamos(genetinės medžiagos pasiskirstymas vyksta atsitiktinai ląstelių dalijimosi metu, to negalima numatyti), ir jo liga pasireiškė visa jėga (tai bus sunki talasemijos forma).
Tik tada mama, tėtis ir abu jų vaikai kreipėsi į gydytoją. Juk prieš tai niekas neturėjo jokių išorinių sveikatos problemų... arba beveik neturėjo.
Dabar su jumis aptarsime, kokius simptomus turėjo šios šeimos nariai, tačiau jie buvo laikomi visai kita liga arba buvo visai nepastebėti.
Kaip pamenate, nesu savidiagnostikos ir savigydos šalininkė, tačiau esu tikra, kad kiekvienai mamai pravartu žinoti keletą požymių, kurie padės laiku kreiptis į tinkamą gydytoją. Tokiu atveju kreipkitės į hematologą!
1. Pirmųjų požymių aptikimo amžius yra toks pat- nuo 6 mėnesių iki 1 metų. Būtent šiuo laikotarpiu šiuolaikiniams vaikams profilaktinių apžiūrų metu atliekami pirmieji kraujo tyrimai. Štai kodėl mūsų pasakojime mama, tėtis ir seneliai iki antrojo vaiko ligos neturėjo diagnozės, nes vaikystėje ne taip dažnai vaikams buvo daromi kraujo tyrimai).
2. Raudonųjų kraujo kūnelių išvaizda dėl abiejų šių problemų labai panašus. Analizės formoje, kurią gaunate laboratorijoje, yra šie elementai: MCV, MCH. Tai raudonųjų kraujo kūnelių rodikliai. Jie apibūdina raudonųjų kraujo kūnelių dydį ir hemoglobino kiekį juose. Tiek talasemijos, tiek geležies trūkumo atveju šie rodikliai, palyginti su norma, labai sumažėja (apie du kartus).
3.Hemoglobino lygis dėl geležies trūkumo sumažintas, iš pradžių, nelabai daug. Taip pat, sergant nedidele talasemija, per pirmuosius 2–3 gyvenimo metus hemoglobino kiekis svyruoja apie apatinę normos ribą, tačiau mažėja nedaug. O iki paauglystės hemoglobinas tampa visiškai normalus, ir tik labai sumažėję eritrocitų rodikliai rodo, kad žmogus yra talasemijos geno nešiotojas.
Kaip matote, pediatras gali labai lengvai nustatyti geležies trūkumą vaikui, sergančiam mažąja talasemija. Šis vaikas daugelį mėnesių bus gydomas geležies preparatais ir nebus išgydytas. Ir tada jis eis pas hematologą. Mano praktikoje tokių atvejų buvo keletas.
1. Gydymo geležies preparatais veiksmingumas: Jei tu jau tris mėnesius, tarkime gerti geležies preparatus ir tuo pačiu nors ir lėtai, bet gerėja vaiko kraujo tyrimai (pirmiausia, padidėja hemoglobino kiekis ir, dar svarbiau, padidėja MCV, MCH indeksai) - tada nesergate talasemija. Gerkite geležį dar porą mėnesių ir visiškai pasveiksite!
2. Tai yra, jei nuoširdžiai jau maždaug 5 mėnesius vartojate geležį kasdien, o jūsų tyrimai nė kiek nepasikeitė į gerąją pusę, tai tikrai yra priežastis pasikonsultuoti su hematologu. Beje, problema gali būti ne talasemija, o prastas individualus geležies įsisavinimas žarnyne. Bet kokiu atveju čia būtinas nuodugnus tyrimas!
3. Kas yra tavo protėviai? Kodėl tai svarbu žinoti? Faktas yra tas, kad talasemija yra labai paplitusi tarp Viduržemio jūros, Pietų ir Pietryčių Azijos bei Afrikos tautų. Rusijoje jis dažniausiai randamas pietiniuose regionuose, ypač Kaukaze. Ir ten pediatrai dažnai greitai nustato teisingą diagnozę. Kadangi jie yra atsargūs dėl paveldimos hemolizinės anemijos. Likusioje Rusijos dalyje talasemija yra labai reta (ja serga apie 1% mūsų šalies gyventojų). Todėl pediatrai pirmiausia pagalvoja apie geležies trūkumą ir jį gydo. O nesėkmės atveju ieško kitų anemijos priežasčių.
Taigi, jei turite bent vieną protėvį iš Kaukazo, o tuo labiau, jei ten gyvenate, būtinai pasidomėkite, ar kuris nors iš jūsų šeimos narių vaikystėje nesirgo nuolatine anemija, ar nebuvo tulžies akmenligės atvejų (kuris yra dažna komplikacija hemolizinė anemija), ar kam buvo pašalinta blužnis ar tulžies pūslė, ar buvo giminaičių, sergančių periodine gelta?
Jei į bet kurį klausimą gausite teigiamą atsakymą arba tiesiog turite rytietiškų šaknų ir nuolatinės, bet lengvos mažakraujystės, būtinai kreipkitės į hematologo konsultaciją.
Kaip jau sakiau, su šia ligos forma išorinių simptomų beveik nėra. Ankstyvame amžiuje vaikas patirs nedidelį hemoglobino sumažėjimą, kuris, kaip taisyklė, nereikalauja gydymo. Jo eritrocitų rodikliai išliks sumažėję visą gyvenimą, o tai nepavojinga sveikatai. Priešingu atveju, toks vaikas, o paskui ir suaugęs žmogus niekuo nesiskiria nuo kitų žmonių. Jis gali būti laikomas talasemijos geno nešiotoju, o ne sergančiu žmogumi. Moterims, turinčioms šį geną, nėštumo metu gali atsinaujinti lengva anemija.
Kodėl aš čia rašau tik apie mažamete talasemija sergančius vaikus? Mat pacientai, sergantys vidutine ir didžiąja formomis, gana sunkiai serga ir dažnai kreipiasi į savo gydantį hematologą. Požiūris į juos visada yra individualus, negali būti bendrų patarimų.
Pacientai, turintys nedidelę formą, pas hematologą kreipiasi kartą per metus, o vėliau – geriausiu atveju, nes jų būklė ypatingų problemų nesukelia. Štai kodėl jiems kyla daugiau klausimų, kurių kartais neturi kam užduoti.
Kaip rašiau pradžioje, esant didžiosioms ir tarpinėms talasemijos formoms, vaikams dažnai reikia perpilti kraują. Dideliuose hematologijos centruose, kur šie pacientai dažnai lankosi, yra individuali donorystės praktika. Tai reiškia, kad kiekvienas vaikas, kuriam reikia reguliariai perpilti kraują, gaus paskiriami keli donorai, kurie paeiliui duoda kraujo tik šiam vaikui.
Kodėl tai daryti? Faktas yra tas, kad jei kiekvieną kartą perpilate vaikui skirtingų donorų kraują, tada po kelių mėnesių tokios terapijos jo imuninė sistema pradeda gaminti antikūnus prieš šiuos svetimus raudonuosius kraujo kūnelius (nors kraujas idealiai tinka grupei ir Rh). veiksnys). Kuo daugiau skirtingų donorų, tuo daugiau antikūnų vaiko kraujyje. Ir tam tikru momentu antikūnų kiekis tampa toks, kad jie pradeda naikinti naują donoro kraujo porciją. Tai yra, paveldimą hemolizinę anemiją taip pat lydi imuninė hemolizinė anemija, po kurios reikia papildomai gydyti vaiką dėl šios būklės.
Kai kūdikis turi penkis ar šešis nuolatinius donorus, ši problema nekyla arba bent jau labai greitai neatsiras.
Be to, tokie planuojamiems donorams reguliariai atliekami visi būtini tyrimai ir mažesnė tikimybė, kad vaikas nuo jų užsikrės rimta infekcija, pvz., ŽIV ar hepatitu.
Taigi, jei turite vyrus ir brolius, kurie nori reguliariai kas kelis mėnesius duoti kraujo (tai nedaro neigiamo poveikio organizmui) tam tikram vaikui, patarkite jiems susisiekti su vietine kraujo perpylimo stotimi arba vaikų hematologijos ligonine. ar tokia būtinybė jūsų mieste.
Pavyzdžiui, aš turiu nuostabų pusbrolį Romą, o jis – nuostabų draugą Antoną, kuris turi tą pačią retą trečiąją kraujo grupę. Abu jie ilgą laiką buvo individualūs donorai vienam iš mano mažųjų pacientų.
Mamos, žinoma, taip pat gali būti planuojamos donorės, bet ne nėštumo būsenoje ir geriau pasibaigus žindymui.
Labai ačiū visiems už dėmesį! Tikiuosi, kad šiandien gauta informacija jums bus naudinga.
25.08.2017
Talasemija priklauso genetinės etiologijos ligų kategorijai. Patologijos procesas grindžiamas tuo, kad raudonieji kraujo kūneliai - reikšmingi kraujo elementai - yra pavojuje. Beta talasemija yra būklė, kai hemoglobinas negamina pakankamai beta baltymų.
Tarp slavų grupės žmonių ši liga beveik nėra paplitusi, jai jautrūs Viduržemio jūros šalių, Afrikos ir Lotynų Amerikos žemynų gyventojai. Dėl šios priežasties daroma prielaida, kad liga veikia kaip tam tikras organizmo apsauginis barjeras nuo maliarijos plazmodio.
Raudonasis kraujo pigmentas hemoglobinas išreiškiamas raudonųjų kraujo kūnelių pavidalu, kurie atlieka svarbų vaidmenį aprūpindami audinius deguonimi per plaučius ir pašalindami iš audinių anglies dioksidą.
Raudonasis kraujo pigmentas hemoglobinas (Hb) išreiškiamas pagrindinių raudonųjų kraujo kūnelių komponentų - raudonųjų kraujo kūnelių, kurie atlieka svarbų vaidmenį aprūpindami audinius deguonimi per plaučius ir pašalindami iš audinių anglies dioksidą, pavidalu. Be to, elementas dalyvauja pašalinant iš organizmo medžiagų apykaitos produktus.
Hemoglobino molekulinė struktūra yra gana glaudžiai susijusi su jo biologine paskirtimi, nes sudėtinga Hb molekulės struktūra tiesiogiai prisideda prie fiziologinių funkcijų vykdymo, įskaitant deguonies transportavimą ir ląstelių mitybą. Be abejo, raudonieji kraujo kūneliai, kurie neša nesveiką hemoglobiną, praranda gebėjimą efektyviai atlikti reikiamą darbą, o tai provokuoja hipochrominės anemijos vystymąsi.
Hemoglobinas žmogaus kraujyje išreiškiamas sudėtingo baltymo, susidedančio iš 4 hemo turinčių subvienetų, įskaitant dvivalenčią geležį, ir atsakingo už gebėjimą aprūpinti kraują deguonimi, pavidalu. Globinas yra baltymas, įtrauktas į Hb molekulinę sudėtį ir glaudžiai susijęs su hemo. Globiną sudaro alfa ir beta grandinių pora, įskaitant aminorūgščių liekanas (α grandinė – 141, β grandinė – 146).
Nemažai nenormalių hemoglobinų susidaro dėl aminorūgščių liekanos pakeitimo vienoje iš globino grandinių, o tai išprovokuoja taškinę mutaciją. Dauguma pakeitimų vyksta β grandinėse; dėl šios priežasties beta talasemija apima plačią hemoglobinopatijų grupę, kurią sukelia sutrikusi Hb molekulės baltyminio komponento gamyba. Tuo pačiu metu α grandinė neturi didelės įvairovės, o tai turi įtakos alfa talasemijos plitimui.

Talasemija yra genetinės kilmės liga, kuri atsiranda, kai nenormalus genas yra paveldimas iš abiejų tėvų ir jo nėra lytinėse chromosomose.
Talasemija yra genetinės kilmės liga, kuri atsiranda, kai nenormalus genas yra paveldimas iš abiejų tėvų ir jo nėra lytinėse chromosomose. Tiesioginis patologijos atsiradimo veiksnys yra įvairūs mutacijų sutrikimai geno, koduojančio vienos ar kitos Hb grandinės sintezę. Patologijos molekulės gali būti pagrįstos defektinės nenormalios mRNR sinteze, struktūriniais genetinės medžiagos pokyčiais, mutacijomis ir neproduktyvia reguliuojančių genų transkripcija. Tokių patologijų rezultatas yra vienos iš polipeptidinių hemoglobino grandinių sintezės sumažėjimas arba nutraukimas.
Taigi, sergant beta talasemija, β grandinių sintezė vyksta nepilname tūryje, todėl susidaro perteklinės α grandinės ir atvirkščiai. Pernelyg susintetintos polipeptidinės grandinės kaupiamos eritrocitų tipo ląstelėse, jas pažeidžiant. Procesą lydi eritrokariocitų sunaikinimas kaulų čiulpuose, eritrocitų hemolizė periferiniame kraujyje ir retikulocitų sunaikinimas blužnyje. Be to, sergant beta talasemija, raudonieji kraujo kūneliai kaupia vaisiaus Hb, kuris neperneša deguonies į audinį, todėl audinių hipoksija. Kaulų čiulpų hiperplazija provokuoja skeleto kaulų deformaciją. Anemija, audinių hipoksija ir neefektyvi eritropoezė didesniu ar mažesniu mastu sukelia vaiko augimo ir vystymosi sutrikimus.
Medicina atpažįsta keletą ligos tipų. Klasifikacija pagrįsta paveiktos polipeptidinės grandinės tipu. Atsižvelgiant į eigos sunkumą, patologija skirstoma į:
Trečioji ligos forma gali baigtis vaiko mirtimi naujagimio laikotarpiu. Pagal metodą ir klasifikaciją medicinos darbuotojai talasemiją išskiria į šiuos tipus:
Be to, patologija dažnai skirstoma į:
Ligą ir jos tipą galima nustatyti vaisiaus vystymosi metu naudojant specialias diagnostikos priemones. Heterozigotinė β-talasemijos forma pasireiškia su nedideliais simptomais arba jų visišku nebuvimu. Jei gydymas atliekamas laiku, apraiškos gali išnykti ir ilgą laiką netrikdyti paciento.
Homozigotinė beta talasemija tampa tiesioginiu nėštumo nutraukimo rodikliu. Dažniausiai vaisiaus mirtis įvyksta prieš gimimą arba kurį laiką po jo (1-2 metai). Tokiais atvejais mirtino Hb kiekis raudonuose pigmentuose smarkiai viršija normalų kiekį.
Patologijos susidarymą palengvina mutacijų procesai 11-osios poros chromosomoje. Šios būklės pacientų hemoglobino kiekis šiek tiek sumažėja. Hematologinis vaizdas apima:
Kai kuriais atvejais nedidelė ligos forma pasireiškia be akivaizdžių fizinių pokyčių. Asmuo nesiskundžia, nors jam gali būti diagnozuota lengva anemija ir mažas raudonųjų kraujo kūnelių kiekis. Genomas apima vieną sveiką ir vieną iš dalies arba visiškai defektuotą. Biocheminiai paciento kraujo tyrimai rodo aukštą geležies, laktatdehidrogenazės ir vaisiaus Gb kiekį.
Klinikinis heterozigotinės talasemijos vaizdas gali nepasireikšti ilgą laiką arba gali būti panašus į kitas ligas, pavyzdžiui:
Medicininės apžiūros metu galite atkreipti dėmesį į:
Vaikams, sergantiems šia patologija, padidėja imlumas infekcijoms, mažėja imunitetas, todėl liga yra sunki.
Suaugusioms moterims nėštumą lydi vaisiaus hidropsijos vystymasis. Pažeidimas nesuderinamas su kūdikio gyvenimu, normalaus gimdymo metu prisideda prie rimtų neurologinių ir psichinių patologijų susidarymo.
Minimali beta talasemija vadinama Silvestroni-Bianco sindromu. Šio tipo liga praeina beveik be apraiškų ir atsitiktinai aptinkama šeimose, kuriose pasireiškia talasemija.
Ši patologijos forma (dar vadinama Cooley anemija) kelia didžiausią pavojų pacientui. Tai sunki anemija, kuri vystosi labai greitai. Nenormalaus proceso simptomai pasireiškia pažodžiui nuo pirmųjų dienų, o aukščiausią lygį pasiekia po 6 mėnesių nuo gimimo. Sergančio vaiko oda blyški su gelta atspalviu.
Be to, būdingi kaukolės ir veido struktūros pokyčiai fiziologiniu lygmeniu, kaulinis audinys nesusiformuoja tinkamai, o tai lydi vystymosi sulėtėjimas.
Pirminiai požymiai yra padidėjęs vidaus organų tūris: kepenys ir blužnis. Organizmą dažnai pažeidžia infekcinės ir uždegiminės ligos, tikėtinas hemoraginio sindromo susidarymas. Endokrininės sistemos veiklos sutrikimai galimi dėl to, kad vaikai nesulaukia brendimo.
Jei Cooley anemija sergančiam pacientui laiku neperpilamas kraujas, gali pasireikšti eritropoetinio audinio hipertrofija. Reguliariai perpylus kraują, prognozė nėra labai gera. Paprastai vidutinė pacientų, sergančių lengva β-talasemija didžiąja forma, gyvenimo trukmė neviršija 20 metų, išskirtiniais atvejais – 30 metų.

Liga gali būti aptikta net nesant klinikinio vaizdo
Medicina pateikia visą spektrą diagnostinių principų, skirtų žmonėms beta talasemijai nustatyti. Liga gali būti aptikta net nesant klinikinio vaizdo. Diagnozės nustatymo pagrindas yra:
Diagnozuodamas gydytojas reikalauja informacijos apie paciento tėvų sveikatos būklę, kad nustatytų genetinių mutacijų tikimybę. Šiuo tikslu taip pat atliekamas kraujo tyrimas dėl DNR. Pagrindinis laboratorinis talasemijos diagnozavimo metodas yra biocheminiai kraujo parametrai, būtent:
Biocheminiai tyrimai atspindi geležies apykaitos sutrikimus, pavyzdžiui, hemolizinės anemijos atveju:
Be laboratorinių kraujo tyrimų, atliekama nemažai kitų tyrimų. Kaulų čiulpų punkcija padeda nustatyti hematopoetinio gemalo tūrį, kraniograma – adatinę periostozę. Pilvo ertmės ultragarsinis tyrimas padeda fiksuoti pakitusius kepenų ir blužnies tūrius. Jei įtariama beta talasemija, gydytojai turi atmesti įvairių tipų anemiją.

Paciento dieta siekiama sumažinti geležies pasisavinimą žarnyne, rekomenduojamos riešutų, sojos, kakavos ir arbatos gėrimų vartojimas.
Beta talasemijos gydymas atliekamas atsižvelgiant į eritropoezės sudėtingumo laipsnį ir genų pažeidimo dėl patologinių procesų lygį. Šiandien naudojami šie metodai:
Bet kokios formos talasemijai reikia vartoti vaistus su folio rūgštimi ir B grupės vitaminais.
Pacientams, kuriems diagnozuota sunki beta talasemija, reikia reguliariai perpilti kraują. Kraujo perpylimas atliekamas nuo pirmųjų vaiko gyvenimo savaičių po gimimo. Tada procedūra pakartojama tam tikru reguliarumu, o tai provokuoja paciento priklausomybės nuo transfuzijos vystymąsi. Kraujo perpylimas yra vienintelis tokio pobūdžio procesas, leidžiantis bent trumpam padidinti Hb kiekį kraujyje. Rekomendacijos dėl kraujo perpylimo yra 95-100 g/l hemoglobino kiekis.

Raudonųjų kraujo kūnelių padidėjimą palengvina šviežiai spaustų burokėlių sulčių gėrimas su šaukštu natūralaus medaus, stikline tris kartus per dieną.
Žinoma, beta talasemijos negalima įveikti jokiais vaistažolių preparatais, tačiau kai kurie metodai gali palengvinti ligos pasireiškimą ir pagerinti paciento savijautą, pavyzdžiui:
Privaloma dieta. Pacientams griežtai draudžiama vartoti alkoholį, riebų maistą, keptą maistą ir greitą maistą.
Tokiems žmonėms ypač vertingi produktai, mažinantys geležies pasisavinimą. Norint išvengti kepenų deformacijos, būtina vartoti hepatoprotektorių.
Raudonųjų kraujo kūnelių perpylimas kartu su medikamentiniu gydymu pagerina paciento gyvenimo kokybę. Nepaisant to, tokia terapija visiškai nepašalina diagnozės, nes ji turi pagalbinį tikslą. Žmonių, sergančių sunkia talasemija, gyvenimas paprastai neviršija 7 metų, jei jiems reguliariai atliekamos kraujo perpylimo procedūros. Skausminga vidaus organų deformacija pasižymi progresavimu, o tai atneša vaikui kančias.

Nėščioms nėščiosioms atliekama visa prenatalinė diagnostika ir genetiniai tyrimai
Beta talasemijos prevencija grindžiama užkirsti kelią paveiktų vaikų pastojimui ir gimimui. Tai galima lengvai padaryti atliekant paprastą kraujo tyrimą ir atrankos procesą. Ligos nešiotojai įspėjami apie grėsmę bendrauti su partneriais, paveiktais tos pačios diagnozės.
Siekiant sumažinti sergančio vaiko riziką, įvairių tipų tyrimams imamas kraujas iš poros, sergančios talasemija. Nėščioms nėščiosioms atliekama visa prenatalinė diagnostika ir genetiniai tyrimai. DNR tyrimais ir gaurelių tyrimais galima nustatyti hemoglobinopatiją nuo nėštumo pradžios.
Siekdama apsaugoti savo vaikus nuo bet kokių ligų, būsimoji mama visada turi stebėti savo sveikatą, o beta talasemijos atveju bus svarbus kruopštus požiūris renkantis partnerį.
Šiandien ši liga laikoma nepagydoma. Lengva beta talasemija sergantis naujagimis gali gyventi visavertį gyvenimą be rimtų pasekmių. Tačiau šiuolaikiniai mokslininkai neturi galimybės tiesiogiai garantuoti šios ligos formos. Visų pirma, poroms, turinčioms homozigotinį paveldimumą, reikia galvoti apie vaikų gimdymą.
Hematologas
Aukštasis išsilavinimas:
Hematologas
Samaros valstybinis medicinos universitetas (SamSMU, KMI)
Išsilavinimo lygis – Specialistas
1993-1999
Papildomas išsilavinimas:
"Hematologija"
Rusijos medicinos magistrantūros akademija
Beta talasemija yra bendras terminas, apimantis daugybę genetiškai nulemtų kraujo sutrikimų. Ši patologinė būklė yra gana reta. Kad atsirastų jam būdingi simptominiai pasireiškimai, abu tėvai turi perduoti sugedusį geną vaikui. Ši patologinė būklė gali pasireikšti arba lengva forma, nesukeliančia akivaizdaus kūdikio sveikatos pablogėjimo, arba ypač sunkia. Šiuo metu ši liga jau gana gerai ištirta. Kai diagnozė patvirtinama, vaiko tėvai turi psichiškai pasiruošti ilgai kovai su šios patologinės būklės apraiškomis.
Verta paminėti, kad dabar ši genetinė liga negali būti visiškai išgydyta, tačiau tinkama palaikomoji terapija gali žymiai pagerinti pacientų kokybę ir gyvenimo trukmę. Šios ligos prognozė priklauso nuo ligos sunkumo. Vaikų, sergančių beta talasemija kūdikystėje ir paauglystėje, mirtingumas yra itin didelis. Taigi, esant sunkioms ligos formoms, reikalingas ilgalaikis medikamentinis gydymas, apimantis daug manipuliacijų, kuriomis siekiama pašalinti esamus sutrikimus.

Ši patologinė būklė pirmą kartą aprašyta 1925 m., kai amerikiečių pediatrai ištyrė emigrantų iš Italijos vaikus. Daugeliu atvejų buvo aptiktas maždaug toks pat vaizdas, įskaitant anemijos požymius, blužnies ir kepenų padidėjimą. Vėliau pasirodė moksliniai darbai, kuriuose buvo aprašyta lengva šios patologinės būklės eiga.
Terminas „talasemija“ buvo įvestas tik 1936 m. Šios ligos pavadinimas pažodžiui verčiamas kaip „šlapios pakrantės liga“. Be to, būtent šiais metais buvo pasiūlyta, kad ši patologinė būklė yra hemoglobino grandinių gamybos sutrikimo pasekmė. Po tyrimų visa medikų bendruomenė sužinojo, kas yra talasemija ir kokias pasekmes sukelia ši patologinė būklė.
Tarp slavų tautų atstovų ši liga yra labai reta. Dažniausiai tai diagnozuojama žmonėms iš Viduržemio jūros šalių, be to, kai kurioms Afrikos tautybėms ir Lotynų amerikiečiams. Šiuo metu egzistuoja teorija, kad tokia mutacija atsirado tam tikroje vietovėje gyvenantiems žmonėms kaip adaptyvus gynybos mechanizmas nuo maliarinio plazmodio. Tačiau kai dviejų tėvų recesyvinė mutacija perduodama vaikui, tai sukelia rimtų pasekmių.
Dabar žinoma, kad sveikų žmonių hemoglobinas susideda iš specialių alfa ir beta grandinių. Kad šis elementas normaliai funkcionuotų, jo gamybą ir tolesnį veikimą turi kontroliuoti genetinio kodo lokusai, įskaitant 11 ir 14 chromosomas. Mutacijos procese šiuose elementuose yra neteisinga informacija. Vėliau tai tampa kraujo ląstelių, turinčių aiškų hemoglobino defektą, gamybos priežastimi. Tai veda prie mirtinų pasekmių.
Šiuo metu žinoma, kad hemoglobinas yra vienintelė žmogaus organizmo medžiaga, galinti surišti deguonies molekules ir pernešti jas iš plaučių į tolimus audinius. Gaminamo hemoglobino struktūros sutrikimas dėl esamos genetinės mutacijos sukelia greitą anemijos vystymąsi, dėl kurios visi audiniai negauna gyvybiškai svarbaus deguonies. Beta talasemija vystosi keliais etapais. Pirma, yra vienos beta grandinės dalies sintezės sutrikimas. Tai sukelia kompensacinius reiškinius. Taigi alfa grandinės frakcijų susidaro perteklius. Dėl spartaus jų daugėjimo pastebimas oksidacinių procesų vystymasis. Alfa grandinių pertekliaus sunaikinimas sukelia raudonųjų kraujo kūnelių membranų sunaikinimą. Raudonųjų kraujo kūnelių struktūros pokyčiai sukelia jų funkcijos sutrikimą.

Dabar žinoma, kad dėl hemoglobino struktūros defekto raudonieji kraujo kūneliai tampa itin jautrūs. Jie greitai subyra, kai liečiasi su kraujagyslių sienelėmis arba kai susiduria vienas su kitu. Iš jų visiškai išsiskiria hemoglobinas. Dėl šio proceso sunaikinami ne tik susidarę raudonieji kraujo kūneliai, bet ir jų pirmtakai, tai yra dar nesubrendę kraujo elementai. Toliau susidaro išsilaisvinantys pigmentai, kurie pirmiausia pradeda prisotinti plazmą, o vėliau nusėda įvairių organų audiniuose. Atsižvelgiant į tai, kad iš sunaikintų raudonųjų kraujo kūnelių išsiskiria daug geležies, šis mikroelementas provokuoja įvairių organų ląstelių sutrikimus ir sukelia rimtų sutrikimų.
Sunkiais atvejais tokių pokyčių pasekmės tampa mirtinos. Visuose gyvybiškai svarbiuose organuose pažeistų ląstelių vietas greitai pakeičia jungiamasis audinys, kuris negali atlikti reikiamų pakeistų vietų funkcijų. Be kita ko, raudonieji kraujo kūneliai, turintys defektą, negali visiškai atlikti savo funkcijos, tai yra, prisotinti organizmo ląsteles deguonimi. Tai tik apsunkina situaciją, todėl patologinių pokyčių sparčiai daugėja. Beveik visų gyvybiškai svarbių organų funkcija palaipsniui mažėja.
Šis terminas yra gana neapibrėžtas ir slepia keletą genetinių ligų grupių, kurias lydi sunkios anemijos formos. Dėl šios priežasties yra daugybė beta talasemijos klasifikavimo būdų. Priklausomai nuo klinikinių apraiškų sunkumo, ši patologinė būklė gali turėti šias formas:
Kai kuriais atvejais ši liga pasireiškia be jokių simptomų. Kitose šalyse per kelias dienas po vaiko gimimo didėjantys sutrikimai sunkios anemijos fone sukelia paciento mirtį. Be kitų dalykų, priklausomai nuo klinikinių apraiškų sunkumo, galima išskirti mažąją, didžiąją ir vidutinę beta talasemiją. Kiekviena forma turi savo būdingus bruožus ir jų vystymosi mechanizmus.
Viena iš šios patologinės būklės klasifikacijų atsižvelgia į sugedusių genų perdavimo vaikui kelią. Pagal šį principą išskiriamos homozigotinės ir heterozigotinės beta talasemijos atmainos. Tai ne visada įmanoma nustatyti. Homozigotinė forma patvirtinama tik tuo atveju, jei mutavęs genas yra abiejuose tėvuose. Šio tipo liga yra labiausiai paplitusi. Heterozigotinė beta talasemija yra paveldima tik iš vieno iš tėvų. Šiuo metu mutavusių genų perdavimo procesai šios ligos vystymosi metu jau yra gerai ištirti.

Daugeliu atvejų abu tėvai yra šio patologinio geno nešiotojai, tačiau patys neserga. Kai susiformuoja vaiko geno kodas, jis mutavusį geną gauna iš abiejų tėvų. Tuo pačiu, jei vienas iš tėvų serga lengva ar net sunkia beta talasemijos forma, o kitas nėra mutavusio geno nešiotojas, rizika susirgti šia patologine vaiko būkle yra minimali. Tokiu atveju vieno iš tėvų sveikas genas visiškai pakeičia mutavusį. Taigi heterozigotinė beta talasemija dabar yra labai reta.
Yra perinataliniai tyrimai, leidžiantys nustatyti šios patologinės būklės buvimą, taip pat jos formą intrauterinio vystymosi laikotarpiu. Homozigotinė talasemija yra nėštumo nutraukimo indikacija. Paprastai vaisius miršta paskutinį trimestrą arba kelias savaites po gimimo.
Tik retais besimptomės eigos atvejais esami požymiai neleidžia nustatyti šios ligos buvimo vaikui ankstyvame amžiuje. Paprastai, kai kalbama apie beta talasemiją, simptomai labai priklauso nuo ligos formos. Taigi kai kuriais atvejais pasireiškimai bus labai lengvi, o kitais - labai sunkūs. Didžioji beta talasemija diagnozuojama gana dažnai. Ši patologijos forma vadinama Cooley karštine, ji yra labai sunki ir lydima rimtų sutrikimų. Daugeliu atvejų simptomai išryškėja jau pirmosiomis kūdikio gyvenimo dienomis. Jie pasiekia savo apogėjų maždaug 6 mėnesius. Būdingi beta talasemijos vystymosi požymiai yra šie:

Daugeliui vaikų, kurie nemiršta per pirmuosius šešis gyvenimo mėnesius, dėl didžiosios beta talasemijos išsivystymo atsiranda rimtų endokrininės sistemos sutrikimų. Gali pasireikšti nykštukiškumas ir uždelstas brendimas. Šios ligos formos prognozė itin nepalanki, net ir dažnai perpylus kraują bei atliekant kitas medicinines procedūras, pacientų gyvenimo trukmė retai viršija 20 metų. Maždaug 30% vaikų miršta pirmaisiais gyvenimo metais. Dauguma pacientų tinkamai gydant išgyvena iki 8 metų.
Be to, beta talasemija yra gana dažna. Šiuo atveju mutacija aptinkama išskirtinai 11-oje chromosomų poroje. Dėl to pacientams pasireiškia tik lengva anemija, hemoglobino kiekis sumažėja tik šiek tiek. Būdingos klinikinės mažosios beta talasemijos apraiškos:
Dažnai ši ligos forma pasireiškia be rimtų fiziologinių pakitimų. Esant mažajai talasemijai, vaikai neturi jokių apraiškų, turinčių įtakos jų bendrajai sveikatai. Galima lengva anemija, nes raudonųjų kraujo kūnelių skaičius yra šiek tiek mažesnis nei įprasta. Be to, kraujo tyrimo rezultatai gali rodyti padidėjusį geležies ir kai kurių kitų medžiagų kiekį, rodantį problemą. Šiuo atveju vaikai vystosi normaliai, tačiau diagnozė yra nepaprastai svarbi, nes ateityje suaugusieji turi planuoti šeimą ir vaikų gimimą, kad jų palikuonys būtų sveiki.
Beta talasemija tarpinė patvirtinama, kai pacientui vieno geno mutacija yra lengva, o kitame – sunki. Šiuo atveju ligos požymiai yra gana neaiškūs, ir jos negalima 100% priskirti nei pagrindinei, nei mažajai beta talasemijos formai. Paprastai vaikų oda pagelsta nuo pirmųjų gyvenimo dienų, tačiau hemoglobino lygis yra ties apatine normos riba. Esant tokiai patologijos eigai, kraujo perpylimas turėtų būti atliekamas gana retai, nes būdingi anemijos požymiai nepastebimi.
Kai kuriais atvejais gali būti nustatytas labai padidėjęs geležies kiekis, o tai yra blužnies pašalinimo indikacija. Pacientą reikia nuolat stebėti gydytojo, nes beta talasemija gali progresuoti į sunkesnę eigą. Sergant tokio tipo patologija, pacientams labai svarbu laikytis sveiko gyvenimo būdo, nes blogi įpročiai gali išprovokuoti būklės pablogėjimą.
Sunkios ligos formos, pavyzdžiui, Cooley anemija, sukelia daugybę sunkių sutrikimų. Dažnos komplikacijos yra hemosiderozė. Šios patologinės būklės, kuri išsivysto beta talasemijos fone, būdingos apraiškos:
Daugumai pacientų vėliau išsivysto didelių sąnarių sinovitas, kuris labai paveikia gebėjimą normaliai judėti. Daugeliu atvejų talasemija yra labai sunki liga. Sutrikęs kaulų augimas skatina lūžius. Be to, dažnos beta talasemijos komplikacijos yra kepenų cirozė, kasos fibrozė, kardiosklerozė, trofinės opos ant kojų, uždegimai plaučiuose, lytinės raidos sutrikimai, didėjantis širdies nepakankamumas, cholecistitas, pneumonija ir sepsis. Be to, gali sutrikti protinis ir fizinis vystymasis.

Daugeliu atvejų būdingi patologinės būklės simptomai gana aiškiai išryškėja jau pirmosiomis kūdikio gyvenimo dienomis, todėl pediatras gali nedelsdamas paskirti konsultacijas su daugeliu specializuotų specialistų ir atlikti reikiamus tyrimus. Liga talasemija dažnai diagnozuojama atliekant genetinius perinatalinius tyrimus, todėl tėvai, anksčiau gavę išsamią informaciją apie vaiko būklę iki jo gimimo, jau yra pasirengę kovoti už jo sveikatą. Kai kuriais atvejais ši patologinė būklė nustatoma pagal požymius, esančius kūdikiui po gimimo. Kalbant apie ligą, tokią kaip talasemija, diagnozė paprastai apima tokius tyrimus kaip:
Įvertinus vaiko būklę, taip pat gavus įvairių tyrimų ir tyrimų duomenis, galima greitai nustatyti esamos ligos pobūdį. Be kita ko, patologinės būklės eigos nustatymas leidžia tiksliai suprasti, kokias pasekmes ji turės.
Požiūris į šios patologinės būklės gydymą labai priklauso nuo jos eigos sunkumo. Visiems pacientams reikalinga speciali dieta, kuria siekiama sumažinti geležies pasisavinimą žarnyne. Esant besimptomei ar nesunkiai eigai, padidinus arbatos, riešutų, sojos, kavos, kakavos suvartojimą, galima gerokai sumažinti šio mikroelemento patekimą į organizmą.
Sergant vidutine ir didžiąja talasemija, reikia reguliariai perpilti kraują. Tokių procedūrų dažnumas priklauso nuo simptomų sunkumo. Perpylus kraują galima pasiekti laikinų rezultatų. Kai kuriais atvejais tokios procedūros yra susijusios su tam tikru šalutiniu poveikiu. Be to, esant patologinei būklei, tokiai kaip talasemija, gydyti reikia vartoti vaistus, kurie padeda pašalinti geležį iš organizmo. Paprastai šiam tikslui naudojamas Desferal. Šis vaistas yra puiki profilaktika siekiant išvengti tokios pavojingos komplikacijos kaip siderosis.
Jei būklė greitai pablogėja dėl hemolizinės krizės, gali būti skiriamas gydymas didelėmis gliukokortikoidų dozėmis. Jei blužnis žymiai padidėja, o geležies lygis nuolat padidėjęs, rekomenduojama sklenektomija. Šią chirurginę intervenciją geriausia atlikti vaikui sulaukus 8-10 metų. Paprastai po blužnies pašalinimo pastebimas reikšmingas būklės pagerėjimas. Tačiau net ir tokia drastiška priemonė leidžia tik tam tikrą laiką pašalinti esamas simptomines apraiškas.
Veiksmingiausias beta talasemijos gydymas yra kaulų čiulpų transplantacija, kurią apsunkina didelė tokių operacijų kaina ir sunku pasirinkti tinkamą donorą. Išsivysčiusiose Europos šalyse veikia vyriausybinės programos ir labdaros fondai, leidžiantys pacientams, sergantiems beta talasemija, gauti reikiamą gydymą. Geriausi donorai yra artimi giminaičiai. Nepaisant didelio teigiamo poveikio, kurį galima pasiekti atliekant kaulų čiulpų transplantaciją, jos įgyvendinimui taikomi tam tikri amžiaus apribojimai. Dauguma gydytojų rekomenduoja šią operaciją vyresniems nei 5 metų vaikams. Tai žymiai padidina palankaus rezultato tikimybę.
Ši genetinė liga reikalauja tikslinio gydymo. Liaudies gynimo priemonės šiuo atveju gali veikti kaip papildomos. Esant tam tikroms šios ligos formoms, jie gali padėti sumažinti simptomų sunkumą. Norint padidinti kraujo kūnelių skaičių, vaikams gali būti rekomenduojama gerti šviežias burokėlių sultis su nedideliu kiekiu medaus. Norint pagerinti paciento būklę, pakanka gerti po 1 stiklinę produkto 3 kartus per dieną.
Be kita ko, gysločių sultys gali duoti tam tikros naudos. Jis turi būti vartojamas 1 šaukštelis. prieš valgant. Kitas liaudies receptas nuo beta talasemijos apraiškų yra gerti šviežių morkų, ridikėlių ir burokėlių sulčių mišinį. Šią priemonę reikia gerti 3 kartus per dieną po 2 valg.
Šiuo metu yra daugybė genetinių ligų perinatalinės diagnostikos metodų. Jei tėvams gresia pavojus, gali būti rekomenduojama atlikti chorioninio gaurelio ar vaisiaus vandenų biopsiją. Tai leidžia nustatyti vaisiaus patologiją ankstyvosiose jo vystymosi stadijose. Jei moteris ar vyras žino, kad yra sugedusio geno nešiotojas arba serga lengva ar besimptome beta talasemija, renkantis partnerį reikia būti labai atsargiems.

Nėštumas turi būti suplanuotas. Prieš pastojant porai reikia atlikti genetinius tyrimus, kad būtų galima apskaičiuoti sergančio vaiko galimybę. Jei nėštumo metu buvo patvirtinta, kad vaisius turi beta talasemiją, kuri vėliau išsivystys sunkia forma, dauguma gydytojų rekomenduoja abortą.
Tėvai gali patys nuspręsti, kaip geriausia elgtis šiuo atveju. Gydytojų rekomendacijos yra visiškai pagrįstos, nes ateityje greičiausiai įvyks persileidimas, kuris vėlesniuose etapuose gali būti trauminis moteriai, arba gims vaikas su didėjančiu organų funkcijos sutrikimu. Talasemija, kuri pasireiškia sunkiais simptomais, daugeliu atvejų yra mirtina.
Talasemija reiškia ligas, kurios, kai jos nustatomos, kalba apie X susietą recesyvinį paveldėjimą, tai yra, talasemija neturi aukšto išsivystymo laipsnio. Tikimybė susidurti yra didesnė tiems vaikams, kurių abu tėvai sirgo šia liga ir perduos nenormalų geną savo palikuonims. Jei tokia problema aktuali tik vienam iš tėvų, tai tikimybė, kad talasemija susirgs ir vaikas, nedidelė.
Sergant talasemija, sutrinka procesai, susiję su polipeptidinių grandinių, kurios yra hemoglobino struktūros dalis, sinteze. Dėl šios priežasties atsiranda specialių raudonųjų kraujo kūnelių, kurie turi šiek tiek kitokią formą ir negali pernešti deguonies į kūno ląsteles ir audinius. Didelis deguonies badas, kurį patiria visi audiniai ir sistemos, sukelia rimtų pasekmių, kurios aiškiai matomos vizualiai.
Talasemija nėra plačiai paplitusi, bet dažniau diagnozuota tam tikruose regionuose kur tikimybė susidurti su maliarija didesnė, tai yra Pietų Azijoje ir Vakarų Afrikoje. Kai kuriose Lotynų Amerikos vietose taip pat užregistruoti talasemijos atvejai. Specialistai šią savybę sieja su specifiniu apsaugos sistemos veikimu. Manoma, kad tokiu būdu gyventojų organizmai gavo pirminį kompensacinį ir apsauginį mechanizmą nuo maliarinio plazmodio – talasemija sergantys pacientai maliarija neserga.
Talasemijai būdingas globino grandinių sutrikimas. Tai gali būti vienos ar kelių grandinių gedimas. Bet kokiu atveju sutrinka svarbaus geležies turinčio baltymo hemoglobino gamyba kraujyje, o normali raudonųjų kraujo kūnelių veikla tampa neįmanoma. Remiantis informacija apie tai, kurios globino grandinės sintezė yra sutrikusi ir kuri chromosomų pora yra už tai atsakinga, išskiriamos kelios ligos grupės ir tipai:
Taip pat yra mišri forma, kuri yra gana reta. Mišrioje formoje pastebimi tiek globinų alfa, tiek beta grandžių sutrikimai. Esant tokioms nesėkmėms, vaisius neturi galimybių išgyventi, o vaikas miršta motinos įsčiose.
Talasemijos priežastis yra tam tikrų probleminių genų paveldėjimas. Jei būsimas vaikas genus su mutacija paveldės ir iš tėčio, ir iš mamos, o tai nutinka gana retai, jis greičiausiai susidurs su liga. Jei pavojingas mutantinis genas yra paslėptas tik vieno iš tėvų chromosomose, kūdikis gali tapti nešiotoju. Esant tokiai situacijai tikimybė susirgti liga yra maža, tačiau tam tikra rizika vis tiek egzistuoja, ir teoriškai ją gydytojai vertina 25–50 proc.
Tačiau dažniausiai nebus pastebėta jokių išorinių apraiškų, o naujagimis bus visiškai sveikas. Ekspertai dar nežino, kas tiksliai lemia tokią genetinę mutaciją.
Kadangi ligos vystymasis yra susijęs su raudonųjų kraujo kūnelių sunaikinimu, išoriniai talasemijos požymiai yra labai pastebimi net naujagimiui - paciento oda yra geltona ar net „kanarėlės“ spalva. Palpuodamas gydytojas dažniausiai tai pastebi kepenys ir blužnis šiek tiek padidėję. Ultragarso rezultatai tai patvirtina. Kepenys tampa didesnės dėl pigmento hemosiderino nusėdimo. Palaipsniui gali mažėti organo dydis, padidėti jo tankis.

Kadangi liga sukelia tam tikrus neigiamus organizmo procesus, sukeliančius audinių hipoksiją, vaikui sutrinka kaulų struktūrų formavimasis, pastebimi fizinio ir psichinio vystymosi sutrikimai. Išorinės talasemijos apraiškos taip pat apima ryškus mongoloidinis veido tipas ir bokšto formos galva. Paprastai viršutinis žandikaulis yra neįprastai didelis. Laikui bėgant, nosies pertvara plečiasi. Laipsniškas žievės kaulų sluoksnio sunaikinimas lems tai, kad maždaug iki pirmųjų gyvenimo metų pabaigos mažo paciento kaulinis audinys pėdų srityje žymiai padidės.
Svarbus ženklas gali būti bendros būklės pablogėjimas lydimas stiprus dusulys. Paprastai dusulys yra susijęs su širdies ir kraujagyslių sistemos būklės pablogėjimu ir galimu širdies nepakankamumu, tačiau, esant talasemijai, širdis ir kraujagyslės gali būti nepriekaištingos, o esant menkiausiam fiziniam poveikiui gali atsirasti dusulys. pastangos. Pacientą taip pat neramina jėgų praradimas, padidėjęs nuovargis, beveik nuolatinis galvos svaigimas, apetito praradimas ir kiti nemalonūs anemijai būdingi simptomai.
Talasemijos diagnozė yra hematologo pareiga. Esant poreikiui, gydytojas tolimesniam darbui gali pasikviesti kitų sričių specialistus, nes neigiamos kraujo ligos pasekmės pastebimos visuose organizmo organuose ir sistemose, o problemos turi būti diferencijuojamos. Diagnozuodami ligą, specialistai skiria:
Ne mažiau svarbus yra dialogas su pacientu, kurio metu galite gauti informacijos apie šeimos istoriją ir galimus genetinius anomalijas.
Nepaisant visų pastangų, genetikai ir farmakologai dar nesugebėjo sukurti veiksmingų metodų, galinčių išgydyti talasemiją. Dažniau naudojama kaulų čiulpų transplantacija, tačiau jo įgyvendinimas yra susijęs su daugybe sunkumų ir rizikų ir ne visada įmanomas. Todėl šiame etape visos gydytojų pastangos apsiriboja simptomų intensyvumo įtaka ir komplikacijų prevencija.

Sergantiesiems lengva talasemija specialaus gydymo nereikia, jų tik prašoma reguliariai tikrintis kraują ir laikytis kai kurių rekomendacijų dėl gyvenimo būdo ir mitybos. Sudėtingesnių talasemijos formų atvejais pacientams skiriami vaistai ir dieta, skirta hemoglobino ir raudonųjų kraujo kūnelių kiekiui didinti. Pacientui taip pat rekomenduojami metodai ir priemonės, kurios gali pašalinti iš organizmo geležies ir uratų perteklių, įskaitant kraujo perpylimą. Kai kuriais atvejais pacientui gali būti patariama pašalinti blužnį.
Net jei mažajam pacientui pavyksta išgyventi, nes jis susiduria su ne tokia pavojinga liga, deja, niekas negali jam garantuoti sveiko gyvenimo. Dažniausiai talasemija sergantys vaikai kenčia nuo sąnarių audinių problemų, jiems diagnozuojami miokardo distrofiniai pokyčiai, diabetas, dažni cirozės atvejai.
Kaip ir apie daugelį paveldimų, genetiškai nulemtų ligų, apie talasemijos prevencijos galimybę kalbėti nereikia.
Mažiems vaikams, ypač naujagimiams, talasemija kelia realią ir labai rimtą grėsmę. Jei naujagimiui diagnozuojama didžioji talasemija, paciento prognozė yra niūri. Jau pirmaisiais gyvenimo metais vaikas gali mirti- sergant šia liga būdingas didelis mirtingumas. Jei kalbame apie lengvesnę talasemijos formą ir aktyvų gydymą, pacientas turi visas galimybes išgyventi.
Radai klaidą? Pasirinkite jį ir paspauskite Ctrl + Enter
Beta talasemija yra nevienalytė ligų grupė, kuriai būdinga sumažėjusi beta globino grandinių sintezė arba jos nebuvimas. Priklausomai nuo būklės sunkumo, yra 3 beta talasemijos formos: pagrindinė, vidutinė ir nedidelė. Klinikinių apraiškų sunkumas yra tiesiogiai proporcingas globino grandinių disbalanso laipsniui. Atsižvelgiant į beta-globino grandinių sintezės sumažėjimo laipsnį, išskiriami:
Beta talasemija yra labiausiai paplitusi talasemijos forma, kurią sukelia sumažėjusi beta grandinių gamyba.
Šis genas paplitęs tarp etninių grupių, gyvenančių Viduržemio jūros baseine, ypač Italijoje, Graikijoje ir Viduržemio jūros salose, taip pat Turkijoje, Indijoje ir Pietryčių Azijoje. Nuo 3 iki 8% italų ar graikų protėvių amerikiečių ir 0,5% juodaodžių amerikiečių yra beta talasemijos geno nešiotojai. Pavienių sporadinių ligos atvejų pasitaiko visuose pasaulio regionuose; tai yra spontaniškai atsirandančios mutacijos arba atvežtos iš vietovių, kuriose yra didelis beta talasemijos geno dažnis. Kai kurie Azerbaidžano ir Gruzijos regionai yra endeminiai talasemijai. Kaip ir pjautuvo ląstelių genas, talasemijos genas yra susijęs su padidėjusiu atsparumu maliarijai, o tai gali paaiškinti geografinį ligos pasiskirstymą.
Beta talasemijos priežastys
Beta talasemiją sukelia 11 chromosomos beta globino lokuso mutacijų serija, pažeidžianti beta globino grandinės sintezę. Buvo aprašyta daugiau nei 100 mutacijų, dėl kurių blokuojami įvairūs genų ekspresijos etapai, įskaitant transkripciją, mRNR apdorojimą ir vertimą. Promotorių mutacijos, ribojančios mRNR transkripciją ir mutacijos, sutrikdančios mRNR susijungimą, paprastai sumažina beta grandinės sintezę (beta + -talasemija), o nesąmoningos mutacijos koduojamoje srityje, sukeliančios priešlaikinį beta globino grandinės sintezės sustabdymą, lemia visišką jų nebuvimą (beta 0 talasemija). .
Beta talasemijos patogenezė
Beta talasemijos patogenezė siejama tiek su nesugebėjimu susintetinti reikiamo kiekio normalaus hemoglobino, tiek su santykinai netirpių α grandinės tetramerų buvimu, kurie susidaro dėl nepakankamo beta grandinių skaičiaus. Dėl neadekvačios hemoglobino sintezės atsiranda hipochrominė mikrocitinė anemija, o dėl nesubalansuoto α-globino grandinių kaupimosi susidaro α 4 tetramerai, kurie nusėda besivystančiose ir brandžiuose eritrocituose. Retikuloendotelinės sistemos ląstelės pašalina tarpląstelines hemoglobino nuosėdas iš eritrocitų, dėl ko pastarieji pažeidžiami, sutrumpėja jų gyvenimo trukmė ir naikinami eritrokariocitai kaulų čiulpuose bei periferinio kraujo retikulocitai ir eritrocitai blužnyje, išsivysto hemolizė. . Sergant beta 0 -galasemija, per didelis vaisiaus hemoglobino (HbF, OC 2 Y 2) kaupimasis eritrocituose. Kai kuriems pacientams taip pat padidėja HbA 2 kiekis (a 2 5 2). HbF turi padidėjusį afinitetą deguoniui, todėl padidėja audinių hipoksija, sutrinka vaiko augimas ir vystymasis. Hemolizė sukelia ryškią eritroidų hiperplaziją ir reikšmingą hematopoetinių zonų tūrio padidėjimą, o tai savo ruožtu sukelia skeleto anomalijas. Neefektyvi eritropoezė (raudonųjų kraujo kūnelių naikinimas kaulų čiulpuose) padidina geležies pasisavinimą, todėl net ir talasemija sergantiems pacientams, kuriems nebuvo perpiltas kraujas, gali išsivystyti patologinis geležies perteklius.
Mažoji beta talasemija
Ji atsiranda dėl vienos beta-talaseminės mutacijos tik vienos chromosomos iš 11 poros. Heterozigotiniams pacientams liga dažniausiai būna besimptomė, hemoglobino kiekis atitinka apatinę normos ribą arba yra šiek tiek sumažėjęs. MCV ir MCH indeksai sumažinami iki tipinio lygio 60-70 fL (normalus - 85-92 fL) ir 20-25 pg (normalus - 27-32 pg).
Hematologinės savybės taip pat apima:
Blužnis padidėja retai ir dažniausiai būna nesunkus.
Hemograma atskleidžia įvairaus sunkumo hipochrominę hiperregeneracinę anemiją. Tipiniais atvejais, prieš koreguojant anemiją perpilant kraują, hemoglobino kiekis yra mažesnis nei 50 g/l. Sergančiųjų talasemija intermedia hemoglobino kiekis be kraujo perpylimo palaikomas 60-80 g/l. Kraujo tepinėlis atskleidžia eritrocitų pipochromiją, mikrocitozę, taip pat daugybę keistos formos fragmentuotų poikilocitų ir tikslinių ląstelių. Periferiniame kraujyje, ypač po splenektomijos, randama daug normocitų (branduolių ląstelių).
Biochemiškai nustatoma netiesioginė hiperbilirubinemija; geležies koncentracijos serume padidėjimas kartu su geležies surišimo gebėjimo serume sumažėjimu. Laktato dehidrogenazės lygis yra padidėjęs, o tai rodo neveiksmingą eritropoezę.
Būdingas biocheminis požymis yra vaisiaus hemoglobino kiekio padidėjimas raudonuosiuose kraujo kūneliuose. Pirmaisiais gyvenimo metais jo lygis viršija 70%, tačiau vaikui augant pradeda mažėti. Hemoglobino A 2 lygis yra maždaug 3%, tačiau HbA 2 ir HbA santykis žymiai padidėja. Pacientams, sergantiems mažąja talasemija, HbF lygis padidinamas iki 2 - 6%, HbA 2 - iki 3,4-7%, o tai turi diagnostinę reikšmę; kai kurių pacientų HbA 2 lygis yra normalus, o HbF – 15–20 % (vadinamasis beta talasemijos variantas, kai vaisiaus hemoglobino kiekis yra aukštas).
Didžioji talasemija (Cooley anemija) yra beta alelio homozigotinė forma (J-talasemija, pasireiškianti sunkios progresuojančios hemolizinės anemijos forma. Didžiosios talasemijos apraiškos dažniausiai prasideda antroje pirmųjų gyvenimo metų pusėje. Pacientas serga sunkia forma odos blyškumas, gelta, sunki mažakraujystė (hemoglobinas - 60-20 g/l, raudonieji kraujo kūneliai - iki 2 x 10 12 / l.) Būdingas sustingimas ir skeleto sistemos pokyčiai, ypač kaukolės kauluose. Pacientams yra deformuota kaukolė, dėl kurios susidaro „anemijos paciento veidas“ Coolie“ - bokštas kaukolė, padidėja viršutinis žandikaulis, atstumas tarp orbitų ir akių mongoloidinė forma, smilkinių ir ilčių išsikišimas su Netinkamas sąkandis. Kaukolės rentgenograma kaukolės sinusų srityje turi būdingą "plaukų ant galo" išvaizdą - "plaukuotos kaukolės" arba "ežio" simptomą, vadinamąją adatinę periostozę. Ilguose vamzdiniuose kauluose išsiplėtusios kaulų čiulpų ertmės, suplonėjęs žievės sluoksnis, dažni patologiniai lūžiai.
Ankstyvieji didžiosios talasemijos požymiai yra reikšmingas blužnies ir kepenų padidėjimas, atsirandantis dėl ekstramedulinės hematopoezės ir hemosiderozės. Išsivysčius hipersplenizmui leuko- ir trombocitopenijos fone, dažnos infekcinės komplikacijos, išsivysto antrinis hemoraginis sindromas.
Vyresnių vaikų augimas vėluoja; Dėl endokrininių sutrikimų jie retai pasiekia brendimą.
Rimta ligos komplikacija yra hemosiderozė. Hemosiderozė ir gelta blyškumo fone sukelia žalsvai rudą odos atspalvį. Kepenų hemosiderozė baigiasi fibroze, kuri kartu su besikeičiančiomis infekcijomis sukelia cirozę. Kasos fibrozę komplikuoja cukrinis diabetas. Miokardo hemosiderozė sukelia širdies nepakankamumo vystymąsi; Tokios būklės kaip perikarditas ir stazinis lėtinis širdies nepakankamumas dažnai sukelia galutinę būseną.
Negydomiems pacientams arba pacientams, kuriems kraujo perpylimas buvo atliktas tik anemijos ir hemolizės paūmėjimo laikotarpiais ir nepakankamai dažnai, atsiranda eritropoetinio audinio hipertrofija, lokalizuota tiek kaulų čiulpuose, tiek už jų ribų. Eritroidinio gemalo ląstelių skaičiaus padidėjimas kaulų čiulpuose nėra tikroji gemalo hiperplazija, o defektinių eritroidinių elementų kaupimosi rezultatas. Jų skaičius padidėja dėl didelio raudonojo gemalo branduolių ląstelių dominavimo, o ne dėl jų brendimo ir diferenciacijos. Kaulų čiulpuose kaupiasi nesugebantys diferencijuoti ir sunaikinamos formos, tai yra, dažniausiai stebima neefektyvi eritropoezė. Plačiau kalbant, neefektyvi eritropoezė suprantama ne tik kaip branduolinių eritroidinių ląstelių intramedulinės lizės procesas, bet ir kaip funkciniu požiūriu defektuotų eritrocitų išsiskyrimas į periferinį kraują, anemija, retikulocitozės stoka.
Nuo nuolatinio kraujo perpylimo priklausomo paciento mirtis dažniausiai įvyksta 2-ąjį gyvenimo dešimtmetį; tik keli iš jų išgyvena 3 dešimtmetį. Remiantis išgyvenamumu, išskiriami trys homozigotinės beta talasemijos sunkumo laipsniai: sunki, vystosi nuo pirmųjų vaiko gyvenimo mėnesių ir greitai baigiasi jo mirtimi; lėtinė, dažniausia ligos forma, kuria serga vaikai gyvena iki 5-8 metų; lengvas, kai pacientai išgyvena iki pilnametystės.
Talasemija tarpinė (beta 0 - ir beta + mutacijų derinys).
Šis terminas reiškia pacientus, kurių klinikinės ligos apraiškos yra vidutinio sunkumo tarp pagrindinės ir nedidelės formos; paprastai pacientai paveldi dvi beta talasemines mutacijas: vieną silpną ir vieną sunkią. Kliniškai stebima gelta ir vidutinio sunkumo splenomegalija, hemoglobino kiekis 70-80 g/l. Sunkios anemijos nebuvimas leidžia nesiimti nuolatinių kraujo perpylimų, tačiau perpylimo terapija juose gali padėti išvengti pastebimų kosmetinių defektų ir kaulų anomalijų. Net ir reguliariai neperpylus šių pacientų organizme išlieka dideli geležies kiekiai, todėl gali išsivystyti hemosiderozė. Dažnai atsiranda splenektomijos indikacijų.

2018 metais gimimo pasninkas prasidės lapkričio 28 d. Šiuo laikotarpiu stačiatikiai ruošiasi švęsti Kalėdas...

Daugumos moterų svajonė yra sukurti šeimą. Jie nori turėti mylintį vyrą ir krūvą vaikų. Bet tai ne visada yra santykiai...

Šiame straipsnyje pateikiama: galingiausia skyrybų malda – informacija, paimta iš viso pasaulio, elektroninio tinklo ir...

Informacinė svetainė apie ikonas, maldas, stačiatikių tradicijas. Malda už skandalus ir kivirčus šeimoje, su vyru, su vaikais...

Kokie būtų Naujieji metai be šampano, mandarinų, Olivier, aspicų ir visų mėgstamo „Silkės po kailiu“. Su paskutiniu...

Paruoškime sausainiams reikalingus ingredientus. Pirmas dalykas, kurį reikia padaryti, yra užvirti vandenį. Mums reikia...

Ar galima registruoti darbuotoją į finansų direktoriaus – vyriausiojo buhalterio pareigas? Vyriausioji buhalterė tvirtina...

Smulkaus verslo vadovas gali lengvai valdyti biudžetą savarankiškai. PATIKRINTA! Jei pavyks...

Naujų projektų kūrimas apima preliminarų ekonominį jų pagrįstumo pagrindimą, vėlesnius...
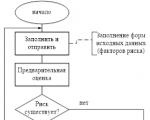
Ataskaitas rengia RM, dėl jų susitaria (patvirtina) Rizikos komitetas prie valdybos ir perduoda...

Didelės, labai didelės pievos pakraštyje, ant ilgo smaragdinio žolės stiebo gyveno mažytė boružėlė. Dievo mažutė...

Šiais laikais gana įprasta, kad žmonės atsigręžia į žvaigždes. Horoskopo pagalba žmogus gali sužinoti...

Verslo ar draugiško. Jei persekiojote nepažįstamą žmogų, tai reiškia jūsų pasitikėjimo moteriškąja lytimi lygį...

pagal Freudo svajonių knygą Jei svajojote, kaip žvejojote, tai reiškia, kad realiame gyvenime vargu ar galite išsijungti...

Daugumos moterų svajonė yra sukurti šeimą. Jie nori turėti mylintį vyrą ir krūvą vaikų. Bet ne visada...

Šiame straipsnyje yra: pati galingiausia malda už skyrybas – informacija paimta iš viso pasaulio, elektroninė...