Kas galima ir ko negalima per Gimimo pasninką?
2018 metais gimimo pasninkas prasidės lapkričio 28 d. Šiuo laikotarpiu stačiatikiai ruošiasi švęsti Kalėdas...
Naujų vaistų kūrimo procesas vykdomas pagal tarptautinius GLP (Good Laboratory Practice), GMP (Good Manufacturing Practice) ir GCP (Good Clinical Practice) standartus.
Klinikiniai vaistų tyrimai apima sistemingą tiriamojo vaisto tyrimą žmonėms, siekiant ištirti jo gydomąjį poveikį arba nustatyti nepageidaujamą reakciją, ir absorbcijos, pasiskirstymo, metabolizmo ir išskyrimo iš organizmo tyrimą, siekiant nustatyti jo veiksmingumą ir saugumą.
Klinikiniai vaisto tyrimai yra būtinas bet kurio naujo vaisto kūrimo etapas arba medikams jau žinomo vaisto vartojimo indikacijų išplėtimas. Pradinėse vaistų kūrimo stadijose atliekami cheminiai, fiziniai, biologiniai, mikrobiologiniai, farmakologiniai, toksikologiniai ir kiti tyrimai su audiniais (in vitro) arba su laboratoriniais gyvūnais. Tai vadinamieji ikiklinikiniai tyrimai, kurių tikslas – gauti mokslinius vaistų veiksmingumo ir saugumo įvertinimus bei įrodymus. Tačiau šie tyrimai negali suteikti patikimos informacijos apie tai, kaip tiriami vaistai veiks žmones, nes laboratorinių gyvūnų organizmas nuo žmogaus skiriasi tiek farmakokinetinėmis savybėmis, tiek organų bei sistemų atsaku į vaistus. Todėl būtina atlikti klinikinius vaistų tyrimus su žmonėmis.
Klinikinis vaistinio preparato tyrimas (testas) – tai sisteminis vaisto tyrimas naudojant jį žmonėms (pacientui ar sveikam savanoriui), siekiant įvertinti jo saugumą ir veiksmingumą, taip pat nustatyti ar patvirtinti jo klinikinį, farmakologinį, farmakodinamines savybes, įvertinti absorbciją, pasiskirstymą, metabolizmą, išsiskyrimą ir sąveiką su kitais vaistais. Sprendimą pradėti klinikinį tyrimą priima užsakovas, atsakingas už tyrimo organizavimą, stebėjimą ir finansavimą. Atsakomybė už praktinį tyrimo atlikimą tenka tyrėjui. Paprastai rėmėjas yra farmacijos įmonė, kurianti vaistus, tačiau tyrėjas taip pat gali veikti kaip rėmėjas, jei tyrimas buvo inicijuotas jo iniciatyva ir jam tenka visa atsakomybė už jo atlikimą.
Klinikiniai tyrimai turi būti atliekami laikantis pagrindinių Helsinkio deklaracijos etikos principų, GСP (Geros klinikinės praktikos) ir taikomų reguliavimo reikalavimų. Prieš pradedant klinikinį tyrimą, reikia įvertinti santykį tarp numatomos rizikos ir laukiamos naudos tiriamajam ir visuomenei. Svarbiausias dalykas yra subjekto teisių, saugos ir sveikatos prioriteto principas prieš mokslo ir visuomenės interesus. Dalykas gali būti įtrauktas į tyrimą tik remiantis savanorišku informuotu sutikimu (IS), gautu išsamiai peržiūrėjus studijų medžiagą. Pacientai (savanoriai), dalyvaujantys tiriant naują vaistą, teisės aktų nustatyta tvarka turi gauti informaciją apie tyrimų esmę ir galimas pasekmes, numatomą vaisto veiksmingumą, rizikos laipsnį, sudaryti gyvybės ir sveikatos draudimo sutartį, ir bandymų metu būti nuolat prižiūrimi kvalifikuoto personalo. Kilus grėsmei paciento sveikatai ar gyvybei, taip pat paciento ar jo teisėto atstovo prašymu, klinikinio tyrimo vadovas privalo sustabdyti tyrimą. Be to, klinikiniai tyrimai sustabdomi, jei vaistas nepasiekiamas arba nepakankamai veiksmingas arba pažeidžiami etikos standartai.
Pirmasis vaisto klinikinių tyrimų etapas atliekamas su 30–50 savanorių. Kitas etapas – išplėstiniai tyrimai 2–5 klinikų pagrindu, kuriuose dalyvauja daug (keli tūkstančiai) pacientų. Kartu pildomos individualios pacientų kortelės, kuriose detaliai aprašomi įvairių tyrimų rezultatai – kraujo tyrimai, šlapimo tyrimai, ultragarsas ir kt.
Kiekvienas vaistas patiria 4 klinikinių tyrimų fazes (etapus).
I fazė. Pirmoji naujos veikliosios medžiagos naudojimo žmonėms patirtis. Dažniausiai studijos pradedamos su savanoriais (sveikų suaugusių vyrų). Pagrindinis tyrimo tikslas – nuspręsti, ar tęsti darbą su nauju vaistu ir, jei įmanoma, nustatyti dozes, kurios bus naudojamos pacientams II fazės klinikinių tyrimų metu. Šio etapo metu mokslininkai gauna preliminarius duomenis apie naujojo vaisto saugumą ir pirmą kartą aprašo jo farmakokinetiką bei farmakodinamiką žmonėms. Kartais I fazės tyrimų su sveikais savanoriais atlikti neįmanoma dėl šio vaisto toksiškumo (gydant vėžį, AIDS). Šiuo atveju neterapiniai tyrimai atliekami, dalyvaujant šia patologija sergantiems pacientams specializuotose įstaigose.
II etapas. Paprastai tai yra pirmoji vaisto vartojimo patirtis pacientams, sergantiems ta liga, kuriai skirtas vaistas. Antroji fazė skirstoma į IIa ir IIb. IIa fazė yra terapiniai bandomieji tyrimai, nes jų rezultatai leidžia optimaliai planuoti tolesnius tyrimus. IIb fazės tyrimai yra didesni tyrimai su pacientais, sergančiais liga, kuri yra pagrindinė naujo vaisto indikacija. Pagrindinis tikslas – įrodyti vaisto veiksmingumą ir saugumą. Šių tyrimų (pagrindinio tyrimo) rezultatai yra III fazės studijų planavimo pagrindas.
III etapas. Daugiacentriniai tyrimai, kuriuose dalyvauja didelės (ir, jei įmanoma, įvairios) pacientų grupės (vidutiniškai 1000–3000 žmonių). Pagrindinis tikslas – gauti papildomų duomenų apie įvairių vaisto formų saugumą ir veiksmingumą, dažniausiai pasitaikančių nepageidaujamų reakcijų pobūdį ir kt. Dažniausiai šios fazės klinikiniai tyrimai yra dvigubai akli, kontroliuojami, atsitiktinių imčių, o tyrimo sąlygos yra kuo artimesnės normaliai įprastai medicinos praktikai. III fazės klinikinių tyrimų metu gauti duomenys yra pagrindas rengiant vaisto vartojimo instrukcijas ir priimant sprendimą dėl jo registravimo Farmakologijos komitete. Rekomendacija klinikiniam naudojimui medicinos praktikoje laikoma pagrįsta, jei naujasis vaistas:
IV etapas. Tyrimai atliekami po vaisto pateikimo į rinką, siekiant gauti išsamesnės informacijos apie ilgalaikį vartojimą įvairiose pacientų grupėse ir su įvairiais rizikos veiksniais ir kt. ir taip visapusiškiau įvertinti narkotikų strategiją. Tyrime dalyvauja daug pacientų, todėl galima nustatyti anksčiau nežinomus ir retus nepageidaujamus reiškinius.
Jei vaistas bus naudojamas naujai indikacijai, kuri dar neregistruota, atliekami papildomi tyrimai, pradedant nuo II fazės. Dažniausiai praktikoje atliekamas atviras tyrimas, kurio metu gydytojas ir pacientas žino gydymo metodą (tiriamą vaistą arba palyginamąjį vaistą).
Tirdamas vienkartiniu metodu pacientas nežino, kokį vaistą vartoja (gali būti placebas), o naudojant dvigubai aklą metodą, to nežino nei pacientas, nei gydytojas, o tik tyrimo vadovas (šiuolaikiniame naujo vaisto klinikiniame tyrime keturios šalys: tyrimo rėmėjas (dažniausiai tai yra vaistų gamybos įmonė), monitorius - pagal sutartį dirbanti tyrimų organizacija, gydytojas tyrėjas, pacientas) . Be to, galimos ir trigubai aklos studijos, kai nei gydytojas, nei pacientas, nei tyrimą organizuojantys ir jo duomenis apdorojantys asmenys nežino konkrečiam pacientui paskirto gydymo.
Jei gydytojai žino, kuris pacientas kokiu vaistu gydomas, jie gali spontaniškai įvertinti gydymą pagal savo pageidavimus ar paaiškinimus. Aklųjų metodų naudojimas padidina klinikinio tyrimo rezultatų patikimumą, pašalina subjektyvių veiksnių įtaką. Jei pacientas žino, kad gauna daug žadantį naują vaistą, gydymo efektas gali būti susijęs su jo nusiraminimu, pasitenkinimu, kad pasiektas geidžiamiausias įmanomas gydymas.
Placebas (lot. placere – mėgti, vertinti) reiškia vaistą, kuris akivaizdžiai neturi jokių gydomųjų savybių.Didysis enciklopedinis žodynas apibrėžia placebą kaip „dozavimo formą, kurioje yra neutralių medžiagų. Naudojamas tiriant įtaigos vaidmenį bet kurios vaistinės medžiagos terapiniam poveikiui, kaip kontrolę tiriant naujų vaistų veiksmingumą. išbandyti kokybišką vaistą
Neigiamas placebo poveikis vadinamas nocebo. Jei pacientas žino, kokį šalutinį poveikį turi vaistas, tada 77% atvejų jis pasireiškia vartojant placebą. Tikėjimas tam tikru poveikiu gali sukelti šalutinį poveikį. Remiantis Pasaulio gydytojų asociacijos komentaru Helsinkio deklaracijos 29 straipsniui, „...placebo vartojimas yra pateisinamas, jei dėl to nepadidėja rizika padaryti rimtą ar negrįžtamą žalą sveikatai...“, y., jei pacientas nelieka be veiksmingo gydymo.
Egzistuoja „visiškai aklų tyrimų“ terminas, kai visos tyrime dalyvaujančios šalys yra apakusios nuo konkrečiam pacientui skiriamo gydymo tipo, kol bus analizuojami rezultatai.
Atsitiktinių imčių kontroliuojami tyrimai yra kokybės standartas atliekant mokslinius gydymo veiksmingumo tyrimus. Tyrimo metu pacientai pirmiausia atrenkami iš didelės tiriamos būklės žmonių populiacijos. Tada šie pacientai atsitiktinai suskirstomi į dvi grupes, suderintas pagal pagrindines prognostines ypatybes. Grupės sudaromos atsitiktine tvarka (atsitiktinių formų nustatymas) naudojant atsitiktinių skaičių lenteles, kuriose kiekvienas skaitmuo arba kiekviena skaitmenų kombinacija turi vienodą pasirinkimo tikimybę. Tai reiškia, kad vienos grupės pacientai vidutiniškai turės tokias pačias savybes kaip ir kitos grupės pacientai. Be to, prieš atsitiktinių imčių skirstymą turėtų būti užtikrinta, kad ligos ypatybės, kurios, kaip žinoma, turi didelę įtaką baigčiai, pasireikštų vienodai dažnai gydymo ir kontrolinėje grupėse. Norėdami tai padaryti, pirmiausia turite suskirstyti pacientus į pogrupius su vienoda prognoze ir tik tada juos atsitiktinai suskirstyti į kiekvieną pogrupį – stratifikuota atsitiktinė atranka. Eksperimentinė grupė (gydymo grupė) gauna intervenciją, kuri, kaip tikimasi, bus naudinga. Kontrolinė grupė (lyginamoji grupė) yra lygiai tomis pačiomis sąlygomis kaip ir pirmoji grupė, išskyrus tai, kad jos pacientai nėra veikiami tiriamos intervencijos.
GOST R 56701-2015
RUSIJOS FEDERACIJOS NACIONALINIS STANDARTAS
MEDICINOS NAUDOJIMO VAISTAI
Rekomendacijos dėl ikiklinikinių saugumo tyrimų planavimo tolesniems klinikiniams tyrimams ir vaistų registracijai
Medicinos reikmėms skirti vaistai. Rekomendacijos dėl neklinikinių saugumo tyrimų atliekant klinikinius tyrimus su žmonėmis ir leidimų prekiauti vaistais
OKS 11.020
11.120.01
Pristatymo data 2016-07-01
Pratarmė
1 PARENGTA Techninio standartizacijos komiteto TC 458 „Vaistų kūrimas, gamyba ir kokybės kontrolė“, remiantis jo paties autentišku 4 dalyje nurodyto dokumento vertimu į rusų kalbą.
2 PRISTATYTA Techninio standartizacijos komiteto TC 458 „Vaistų kūrimas, gamyba ir kokybės kontrolė“
3 PATVIRTINTA IR ĮSIgaliojo Federalinės techninio reguliavimo ir metrologijos agentūros įsakymu, 2015 m. lapkričio 11 d. N 1762-st.
4 Šis standartas yra identiškas tarptautiniam dokumentui ICH M3(R2):2009* „Neklinikinių saugumo tyrimų, skirtų klinikiniams tyrimams su žmonėmis atlikti, ir vaistų rinkodaros teisės suteikimo gairės“). Šio standarto pavadinimas buvo pakeistas, palyginti su nurodyto tarptautinio dokumento pavadinimu, kad atitiktų pavadinimus, priimtus esamame standartų rinkinyje „Medicininiai vaistai“. Taikant šį standartą, vietoj referencinių tarptautinių standartų rekomenduojama naudoti atitinkamus Rusijos Federacijos nacionalinius standartus, nurodytus DA priede.
________________
* Prieigą prie tekste minimų tarptautinių ir užsienio dokumentų galite gauti susisiekę su klientų aptarnavimo tarnyba. - Duomenų bazės gamintojo pastaba.
5 PRISTATYTA PIRMĄ KARTĄ
Šio standarto taikymo taisyklės yra nustatytos GOST R 1.0-2012 (8 skyrius). Informacija apie šio standarto pakeitimus skelbiama metiniame (einamųjų metų sausio 1 d.) informaciniame rodyklėje „Nacionaliniai standartai“, o oficialus pakeitimų ir pakeitimų tekstas skelbiamas mėnesinėje informacijos rodyklėje „Nacionaliniai standartai“. Šio standarto peržiūros (pakeitimo) ar panaikinimo atveju atitinkamas pranešimas bus paskelbtas kitame mėnesinio informacijos rodyklės „Nacionaliniai standartai“ numeryje. Atitinkama informacija, pranešimai ir tekstai taip pat skelbiami viešojoje informacinėje sistemoje - oficialioje Federalinės techninio reguliavimo ir metrologijos agentūros svetainėje internete (www.gost.ru).
Įvadas
Šio standarto tikslas – nustatyti bendrus metodus planuojant ikiklinikinius vaistinių preparatų tyrimus su Europos Sąjungos šalimis, Jungtinėmis Amerikos Valstijomis, Japonija ir kitomis šalimis, kurios taiko tarptautines ICH gaires, pagrindžiančias galimybę atlikti klinikinius tam tikras pobūdis ir trukmė, taip pat vėlesnė valstybinė registracija.
Standartas skatina laiku atlikti klinikinius tyrimus, mažinant laboratorinių gyvūnų naudojimą pagal 3R principą (sumažinti/patobulinti/pakeisti), mažinant kitų išteklių naudojimą kuriant vaistus. Reikėtų apsvarstyti naujus alternatyvius metodus in vitro saugos įvertinimui. Jei šie metodai yra tinkamai patvirtinti ir priimti visų ICH gaires įgyvendinančių šalių reguliavimo institucijų, jie gali būti naudojami pakeisti esamus standartinius metodus.
Šis standartas skatina saugų, etišką vaistų kūrimą ir jų prieinamumą pacientams.
Ikiklinikinį saugumo vertinimą, atliekamą valstybinės vaistų registracijos tikslais, paprastai sudaro šie etapai: farmakologiniai tyrimai, bendrieji toksikologiniai tyrimai, toksikokinetiniai ir ikiklinikiniai farmakokinetikos tyrimai, toksiškumo reprodukcijai tyrimai, genotoksiškumo tyrimai. Vaistų, turinčių tam tikrų savybių arba skirtų ilgalaikiam vartojimui, kancerogeniškumo įvertinimas taip pat būtinas. Kitų ikiklinikinių tyrimų poreikis, siekiant įvertinti fototoksiškumą, imunotoksiškumą, toksiškumą nesubrendusiems gyvūnams ir priklausomybės nuo vaistų atsiradimą, nustatomas individualiai. Šis standartas nurodo neklinikinių tyrimų poreikį ir jų ryšį su vėlesniais klinikiniais tyrimais su žmonėmis.
Iki šiol šalys, taikančios ICH gaires, padarė didelę pažangą derindamos šiame standarte aprašytų vaistinių preparatų klinikinių tyrimų neklinikinių saugumo tyrimų laiką. Tačiau kai kuriose srityse skirtumai išlieka. Reguliavimo institucijos ir gamintojai toliau peržiūri šiuos skirtumus ir stengiasi toliau tobulinti vaistų kūrimo procesą.
Šis standartas nustato rekomendacijas dėl neklinikinių saugumo tyrimų planavimo tolesnių klinikinių tyrimų ir vaistinių preparatų registravimo tikslais.
Šis standartas taikomas visais vaistų kūrimo atvejais ir pateikia bendrąsias vaistų kūrimo gaires.
Biotechnologiniais metodais gautiems vaistiniams preparatams turi būti atlikti atitinkami saugumo tyrimai pagal ICH S6 gairę dėl neklinikinių biotechnologinių vaistinių preparatų tyrimų. Šiems vaistams šis standartas taikomas tik ikiklinikinių tyrimų tvarkai, atsižvelgiant į klinikinės raidos fazę.
Siekiant optimizuoti ir paspartinti vaistų, skirtų gyvybei pavojingoms ar sunkioms ligoms (pavyzdžiui, išplitusiam vėžiui, nuolatinei ŽIV infekcijai, įgimtų fermentų trūkumo sąlygotoms ligoms), kurioms šiuo metu nėra veiksmingo gydymo, gydyti, kūrimą, taip pat taikomas individualus požiūris. naudojami toksikologiniam vertinimui ir klinikinei plėtrai. Tokiais atvejais, kaip ir naujoviškų terapinių priemonių (pvz., mažų trukdančių RNR) ir vakcinų adjuvantų, tam tikri tyrimai gali būti sutrumpinti, modifikuoti, pridėti arba ištrinti. Jei yra atskirų farmakoterapinių vaistinių preparatų grupių ICH rekomendacijos, reikia vadovautis pastarosiomis.
Vaistų kūrimas yra žingsnis po žingsnio procesas, apimantis duomenų apie jų veiksmingumą ir saugumą tiek gyvūnams, tiek žmonėms įvertinimą. Pagrindiniai ikiklinikinio vaistų saugumo vertinimo tikslai apima toksiškumo tiksliniam organui, jo dozės ir atsako santykio, santykio su poveikiu (sisteminiu poveikiu) ir, jei taikoma, galimo toksinio poveikio grįžtamumo nustatymas. Šie duomenys naudojami klinikinių tyrimų pradinei saugiai dozei ir dozių diapazonui nustatyti bei galimo nepageidaujamo poveikio klinikinio stebėjimo parametrams nustatyti. Ikiklinikinių saugumo tyrimų, nors klinikinės plėtros pradžioje jų pobūdis yra ribotas, turėtų pakakti, kad būtų parodytas galimas nepageidaujamas poveikis, kuris gali pasireikšti planuojamo klinikinio tyrimo metu.
Klinikiniai tyrimai atliekami siekiant ištirti vaisto veiksmingumą ir saugumą, pradedant santykinai maža sistemine ekspozicija nedaugeliui asmenų. Vėlesniuose klinikiniuose tyrimuose vaisto ekspozicija padidėja ilginant vartojimo trukmę ir (arba) tiriamosios populiacijos dydį. Klinikiniai tyrimai turėtų būti papildyti tinkamais saugumo įrodymais, remiantis anksčiau atliktų klinikinių tyrimų rezultatais ir papildomais neklinikiniais saugumo duomenimis, kurie gaunami progresuojant klinikinei plėtrai.
Klinikiniai arba ikiklinikiniai duomenys apie sunkų nepageidaujamą poveikį gali turėti įtakos klinikinių tyrimų tęsimui. Šie duomenys turėtų būti peržiūrėti kaip bendro klinikinio vystymosi plano dalis, siekiant nustatyti papildomų ikiklinikinių ir (arba) klinikinių tyrimų atlikimo ir planavimo galimybes.
Klinikiniai tyrimai atliekami etapais, kurie įvairiose šalyse turi skirtingus pavadinimus. Šiame standarte vartojama terminija, vartojama ICH E8 rekomendacijose dėl bendrųjų principų, taikomų atliekant klinikinius vaistinių preparatų tyrimus. Tačiau, kadangi pastebima ryški klinikinio vystymosi etapų sujungimo tendencija, šiame dokumente tam tikrais atvejais taip pat nurodomas ikiklinikinių tyrimų ryšys su klinikinių tyrimų trukme ir dydžiu, taip pat tiriamųjų charakteristikos ( tikslinę populiaciją).
Neklinikinių saugumo tyrimų ir klinikinių tyrimų su žmonėmis planavimas ir planavimas turėtų būti grindžiamas moksliniais principais ir atitikti etikos principus.
2.1. Didelių dozių parinkimas bendram toksiškumui tirti
Galimas kliniškai reikšmingas poveikis toksikologinių tyrimų metu paprastai gali būti visiškai ištirtas naudojant dozes, artimas maksimaliai toleruojamai dozei (MTD). Tačiau nebūtina patvirtinti MTD kiekviename tyrime. Taip pat leidžiama naudoti ribotas dideles dozes, įskaitant dozes, kurios kartojasi dozes, kurios, kaip tikimasi, bus naudojamos klinikinėje praktikoje (klinikinė ekspozicija) arba kai pasiekiama didžiausia pasiekiama ekspozicija (sotumo ekspozicija) arba priimtina didžiausia dozė (MFD). Naudojant šias ribotas dideles dozes (išsamiau aprašyta toliau ir 1 paveiksle), gyvūnams neskiriama dozių, kurios nesuteikia papildomos informacijos klinikiniam saugumui numatyti. Šis metodas atitinka panašias rekomendacijas dėl toksiškumo reprodukcijai ir kancerogeniškumo tyrimų planavimo, pagal kuriuos jau nustatytos ribotos didelės dozės ir (arba) poveikis.
Ribota didelė 1000 mg/kg per parą dozė, skirta ūminio, polėtinio ir lėtinio toksiškumo tyrimams su graužikais ir negraužkais, yra tinkama visoms reikmėms, išskyrus tuos, kurie aptariami toliau. Kai kuriais atvejais, kai 1000 mg/kg per parą dozė neužtikrina 10 kartų didesnės nei klinikinės ekspozicijos, o klinikinė vaisto dozė viršija 1 g per parą, toksikologinių tyrimų metu dozės turėtų būti apribotos iki 10 kartų didesnės už dozę. pasiekti klinikinę ekspoziciją, 2000 mg /kg per parą dozę arba naudoti MFD, pasirenkant mažiausią. Tais retais atvejais, kai 2000 mg/kg per parą dozė yra mažesnė už klinikinę ekspoziciją, gali būti naudojama didesnė dozė iki MFD.
Dozės, kurios suteikia 50 kartų didesnę sisteminę ekspoziciją (paprastai nustatomos pagal pradinės medžiagos arba farmakologiškai aktyvios provaisto molekulės vidutines AUC vertes (1 pastaba)), palyginti su sistemine klinikine ekspozicija, taip pat laikomos priimtinomis didžiausiomis dozėmis esant ūminiam ir ūminiam toksiškumui. pakartotinis vartojimas bet kuriai gyvūnų rūšiai.
Norint pradėti III fazės klinikinius tyrimus Jungtinėse Amerikos Valstijose, toksikologiniai tyrimai, naudojant ribotas dideles dozes, atliekami su mažiausiai vienos rūšies gyvūnais, kurių dozė yra 50 kartų didesnė už ekspoziciją. Jei toks metodas neįmanomas, tyrimą rekomenduojama atlikti su vienos rūšies gyvūnais 1 mėnesį ar ilgiau, naudojant ribotą didelę 1000 mg/kg dozę, MFD arba MTD, atsižvelgiant į tai, kuri yra mažesnė. Tačiau tam tikrais atvejais toks tyrimas gali būti nereikalingas, jei atliekant trumpesnės trukmės tyrimą toksinis poveikis buvo pastebėtas vartojant dozes, didesnes nei 50 kartų už ekspoziciją. Jei genotoksiškumo vertinamieji parametrai yra įtraukti į bendrą toksiškumo tyrimą, atitinkama didžiausia dozė turi būti pagrįsta MFD, MTD arba ribota didele 1000 mg/kg per parą doze.
1 pastaba. Šiame dokumente „ekspozicija“ paprastai reiškia vidutinę AUC vertę grupėje. Kai kuriais atvejais (pavyzdžiui, jei junginys arba junginių klasė gali sukelti ūmius širdies ir kraujagyslių sistemos pokyčius arba simptomai yra susiję su poveikiu centrinei nervų sistemai), tikslingiau poveikio ribas nustatyti naudojant vidutines C grupės vertes.
Farmakologinio ir farmakodinaminio saugumo tyrimai apibrėžti ICH gairėse S7A.
Pagrindinis farmakologinio saugumo tyrimų rinkinys apima poveikio širdies ir kraujagyslių, centrinei nervų ir kvėpavimo sistemoms vertinimą. Paprastai šie tyrimai turėtų būti atliekami prieš klinikinį vystymąsi pagal ICH gairėse S7A ir S7B nustatytus principus, skirtus vaistinių preparatų farmakologiniam saugumui tirti ir ikiklinikiniam žmonėms skirtų vaistinių preparatų gebėjimo sulėtinti skilvelių veiklą įvertinimui. repoliarizacija (pailgina QT intervalą). Jei reikia, klinikinio vystymosi pabaigoje gali būti atliekami papildomi ir tolesni farmakologinio saugumo tyrimai. Siekiant sumažinti laboratorinių gyvūnų naudojimą, kai tik įmanoma, į bendruosius toksiškumo tyrimų protokolus turėtų būti įtraukti kiti vertinimai. in vivo kaip papildomas.
Pirminių farmakodinaminių tyrimų tikslas ( in vivo ir/arba in vitro) yra nustatyti veikliosios medžiagos veikimo mechanizmą ir (arba) farmakologinį poveikį, susijusį su jos siūlomu terapiniu vartojimu. Tokie tyrimai paprastai atliekami ankstyvoje farmacijos kūrimo stadijoje, todėl paprastai neatliekami laikantis geros laboratorinės praktikos (GLP) principų. Šių tyrimų rezultatai gali būti naudojami renkantis dozę tiek ikiklinikiniams, tiek klinikiniams tyrimams.
Prieš pradedant klinikinius tyrimus, apskritai reikia įvertinti gyvūnų ir žmonių metabolinį profilį ir prisijungimo prie plazmos baltymų laipsnį. in vitro, taip pat sisteminio poveikio duomenis (ICH S3A Toksikokinetikos tyrimų vadovas) gyvūnų rūšims, naudojamoms kartotinių dozių toksikologijos tyrimuose. Tiriamų rūšių farmakokinetikos (PK) duomenis (t. y. absorbciją, pasiskirstymą, metabolizmą ir išskyrimą) reikia gauti prieš pradedant klinikinius tyrimus su dideliu skaičiumi tiriamųjų arba ilgą laiką (paprastai prieš prasidedant fazei). III klinikiniai tyrimai).gyvūnai ir gauti biocheminiai duomenys in vitro, reikšmingas nustatant galimą vaistų sąveiką. Šie duomenys naudojami žmonių ir gyvūnų metabolitų palyginimui ir papildomų tyrimų poreikiui nustatyti.
Ikiklinikinis metabolito (-ų) apibūdinimas žmonėms yra būtinas tik tada, kai metabolito (-ų) ekspozicija viršija 10 % visos vaisto ekspozicijos, o ekspozicijos mastas žmonėms yra žymiai didesnis nei stebimas toksikologinių tyrimų metu. Tokie tyrimai turi būti atliekami norint gauti patvirtinimą III fazės klinikiniams tyrimams. Vaistams, kurių paros dozė neviršija 10 mg, tokių tyrimų gali prireikti esant didesnei metabolitų proporcijai. Kai kurių metabolitų toksikologiniai tyrimai neatliekami (pvz., dauguma metionino konjugatų) ir jų tyrimų nereikia. Kiekvienu konkrečiu atveju reikia apsvarstyti galimybę atlikti ikiklinikinius metabolitų, kurie gali turėti toksikologinį poveikį (pvz., tik žmonėms būdingą metabolitą), tyrimą.
Tradiciškai ūmaus toksiškumo duomenys buvo gauti atlikus vienos dozės toksiškumo tyrimus su dviem žinduolių rūšimis, naudojant kliniškai siūlomus ir parenterinius vartojimo būdus. Tačiau šią informaciją taip pat galima gauti atlikus tinkamai atliktus dozės didinimo tyrimus arba trumpalaikius dozių diapazono tyrimus, kurių metu nustatomas MTD gyvūnams, naudojamiems atliekant bendruosius toksiškumo tyrimus.
Jei informacijos apie ūmų toksiškumą galima gauti iš kitų tyrimų, atskirų vienos dozės tyrimų nerekomenduojama. Tyrimai, kuriuose pateikiama informacija apie ūmų toksiškumą, gali būti taikomi tik klinikiniam naudojimui siūlomu vartojimo būdu ir negali būti atliekami pagal GLP reikalavimus, jei kartotinių dozių toksiškumo tyrimuose, atliktuose pagal GLP reikalavimus, buvo naudojamas vaisto vartojimo būdas. siūlomas klinikiniam naudojimui. Mirtingumas neturėtų būti privalomas galutinis taškas atliekant ūmaus toksiškumo tyrimus. Kai kuriais ypatingais atvejais (pvz., mikrodozių tyrimai, žr. 7 skyrių), ūmaus toksiškumo tyrimai arba vienos dozės tyrimai gali būti pagrindinis klinikinių tyrimų su žmonėmis priežastis. Tokiais atvejais didelės dozės pasirinkimas gali skirtis nuo aprašyto 1.1 skyriuje, tačiau reikia atsižvelgti į numatomą klinikinę vaisto dozę ir vartojimo būdą. Šie tyrimai turi būti atliekami pagal GLP reikalavimus.
Informacija apie ūmų vaistų toksiškumą gali būti naudojama prognozuojant perdozavimo pasekmes žmonėms ir turėtų būti prieinama prieš pradedant III fazės klinikinius tyrimus. Ambulatorinių klinikinių tyrimų metu gali prireikti anksčiau įvertinti ūminį toksiškumą vaistams, siūlomiems gydyti pacientų, kuriems yra didelė perdozavimo rizika (pvz., depresija, skausmas, demencija).
Rekomenduojama kartotinių dozių toksiškumo tyrimų trukmė priklauso nuo planuojamo tolesnio klinikinio tyrimo trukmės, indikacijos ir tikslo. Apskritai toksiškumo su gyvūnais tyrimų su dviem gyvūnų rūšimis (viena iš jų nėra graužikai) trukmė turėtų būti lygi arba ilgesnė už planuojamą klinikinių tyrimų trukmę iki rekomenduojamos maksimalios kartotinių dozių toksiškumo tyrimų trukmės (1 lentelė). . Ribotos didelės dozės/poveikis, laikomi tinkamais kartotinių dozių toksiškumo tyrimams, aprašyti 2.1.
Tais atvejais, kai klinikinių tyrimų metu pastebimas reikšmingas terapinis poveikis, tyrimo trukmė gali būti pailginta individualiai, palyginti su kartotinių dozių toksiškumo tyrimų trukme, naudojama kaip klinikinių tyrimų pagrindas.
6.1 Klinikinei plėtrai reikalingi tyrimai
Paprastai kartotinių dozių toksiškumo tyrimas su dviem rūšimis (viena iš jų nėra graužikai), trunkantis mažiausiai dvi savaites, yra pakankamas bet kokių iki dviejų savaičių trukmės klinikinių tyrimų pagrįstumui pagrįsti (1 lentelė). Norint pagrįsti ilgesnės trukmės klinikinius tyrimus, reikia atlikti bent tokios pat trukmės toksiškumo tyrimus. Norint pagrįsti ilgesnius nei 6 mėnesius trukusius klinikinius tyrimus, būtinas 6 mėnesių trukmės tyrimas su graužikais ir 9 mėnesių trukmės tyrimas su graužikais (išimtis rasite 1 lentelės pastabose).
1 lentelė. Rekomenduojama kartotinių dozių toksikologinių tyrimų trukmė, reikalinga klinikiniams tyrimams pagrįsti
Maksimali klinikinio tyrimo trukmė | ||
Graužikai | Ne graužikai |
|
Iki dviejų savaičių | 2 savaitės | |
Nuo dviejų savaičių iki šešių mėnesių | Tas pats, kas klinikiniuose tyrimuose |
|
Daugiau nei šeši mėnesiai | 6 mėnesiai | 9 mėn |
Jungtinėse Valstijose vienos dozės išplėstinį toksiškumo tyrimą leidžiama naudoti kaip alternatyvą 2 savaičių trukmės tyrimams, siekiant paremti vienos dozės klinikinius tyrimus (žr. c pastabą 3 lentelėje). Trumpesni nei 14 dienų klinikiniai tyrimai gali būti pateisinami tokios pat trukmės toksiškumo tyrimais. Kai kuriais atvejais klinikiniai tyrimai, trunkantys ilgiau nei 3 mėnesius, gali būti pradėti remiantis 3 mėnesių trukmės tyrimų su graužikais ir ne graužikais rezultatais, jei baigtų lėtinio toksiškumo tyrimų su graužikais ir ne graužikais rezultatai atitinka nacionalinius teisės aktų reikalavimus. klinikiniai tyrimai gali būti pateikti prieš klinikiniam vaistinio preparato vartojimui viršijant 3 mėnesius. Sunkių ar gyvybei pavojingų ligų atveju arba kiekvienu konkrečiu atveju toks pratęsimas galimas, jei yra visiškai užbaigtų lėtinio toksiškumo tyrimų su graužikais rezultatais ir intravitalinių tyrimų bei nekropsijos tyrimų su graužikais rezultatais. Išsamūs negraužikų patologiniai duomenys turėtų būti gauti per ateinančius 3 mėnesius. Gali būti atvejų, kai vaistas skirtas naudoti pediatrijoje, o turimi ikiklinikiniai tyrimai su gyvūnais (toksikologiniai ar farmakologiniai) rodo galimą poveikį tikslinių organų vystymuisi. Tokiais atvejais gali prireikti ilgalaikių toksiškumo tyrimų su nesubrendusiais gyvūnais (žr. 12 skyrių). Europos Sąjungoje manoma, kad pakanka 6 mėnesių trukmės toksikologinių tyrimų su ne graužikais. Tačiau jei buvo atlikti ilgesnės trukmės tyrimai, papildomi tyrimai, truksiantys ilgiau nei 6 mėnesius, nepriimtini. Toliau pateikiami pavyzdžiai, kai 6 mėnesių trukmės tyrimai be graužikų taip pat tinka klinikiniams tyrimams Japonijoje ir JAV paremti: Jei imunogeniškumas ar netoleravimas neleidžia atlikti ilgalaikių tyrimų; Trumpalaikiam poveikiui pakartotinai vartojant, net jei klinikinio tyrimo trukmė viršija 6 mėnesius, pavyzdžiui, nereguliariai vartojant migrenos, erekcijos disfunkcijos ar paprastosios pūslelinės atveju; Vaistai, vartojami ilgai, siekiant sumažinti vėžio pasikartojimo riziką; Vaistai, vartojami esant indikacijoms, kurioms nustatyta trumpa gyvenimo trukmė. |
||
6.2 Valstybinė registracija
Atsižvelgiant į didelį rizikos grupei priklausančių pacientų skaičių ir santykinai mažiau kontroliuojamas narkotikų vartojimo sąlygas medicinos praktikoje, priešingai nei atliekant klinikinius tyrimus, siekiant pagrįsti galimybę vartoti vaistą medicinoje, ikiklinikiniai tyrimai yra reikalingi ilgesniam laikui, nei pagrįsti klinikinius tyrimus. studijos. Kartotinių dozių toksiškumo tyrimų, kurių reikia norint pagrįsti skirtingo gydymo trukmės vaistinių preparatų vartojimo medicinoje leidimą, trukmė pateikta 2 lentelėje. Kai kuriais atvejais, esant nedideliam skaičiui patologinių būklių, kai rekomenduojama vaisto vartojimo trukmė yra nuo 2 sav. iki 3 mėnesių, tačiau yra didelė klinikinė patirtis, rodanti platesnį ir ilgalaikį klinikinį vartojimą (pvz., nerimas, sezoninis alerginis rinitas, skausmas), toksikologinius tyrimus, kurių trukmė tinkamesnė tais atvejais, kai rekomenduojama narkotikų vartojimo trukmė viršija 3 mėnesius. gali prireikti.
2 lentelė. Rekomenduojama toksikologinių tyrimų trukmė su pakartotiniu skyrimu, reikalinga valstybinei vaistinio preparato registracijai*
Vartojimo trukmė pagal indikacijas | Ne graužikai |
|
Iki dviejų savaičių | ||
Nuo dviejų savaičių iki vieno mėnesio | ||
Nuo vieno mėnesio iki trijų mėnesių | 6 mėnesiai | 6 mėnesiai |
Per tris mėnesius | 6 mėnesiai | 9 mėn |
* Paaiškinimai pateikti 1 lentelės pastabose. |
||
Pirmą kartą žmonėms skirtos dozės nustatymas yra svarbus elementas siekiant užtikrinti ankstyvuosiuose klinikiniuose tyrimuose dalyvaujančių asmenų saugumą. Nustatant rekomenduojamą pradinę dozę žmonėms, reikia įvertinti visus svarbius neklinikinius duomenis, įskaitant farmakologinį dozės ir atsako poveikį, farmakologinį/toksikologinį profilį ir farmakokinetikos duomenis.
Apskritai, svarbiausią informaciją teikia didelė netoksiška dozė (NOAEL), nustatyta atliekant neklinikinius saugumo tyrimus su tinkamiausiomis gyvūnų rūšimis. Apskaičiuota pradinė klinikinė dozė taip pat gali priklausyti nuo įvairių veiksnių, įskaitant farmakodinaminius parametrus, individualias veikliosios medžiagos savybes ir klinikinių tyrimų planą. Atrinkti metodai pateikiami nacionalinėse gairėse.
Tiriamieji klinikiniai tyrimai (8 skirsnis) su žmonėmis gali būti pradėti atliekant mažesnį arba kitokį neklinikinių tyrimų kiekį, nei reikia klinikinio vystymosi tyrimams (6.1), todėl klinikinės pradinės (ir didžiausios) dozės nustatymas gali skirtis. Rekomenduojami pradinių dozių parinkimo kriterijai įvairiuose tiriamuosiuose tyrimuose pateikti 3 lentelėje.
Kai kuriais atvejais ankstyvi duomenys apie žmones gali padėti geriau suprasti fiziologines ir (arba) farmakologines vaisto savybes žmonėms, kuriamo vaisto savybes ir terapinių taikinių svarbą konkrečiai ligai. Racionalūs ankstyvieji tiriamieji tyrimai gali išspręsti tokias problemas. Šiame standarte tiriamieji klinikiniai tyrimai apibrėžiami kaip tyrimai, atlikti ankstyvoje I fazėje, apimantys ribotą ekspoziciją ir nevertinant terapinio veiksmingumo bei klinikinio toleravimo. Jie atliekami tiriant įvairius parametrus, tokius kaip PD, vaisto PK ir kiti biomarkeriai, kurie gali apimti receptorių prisijungimą ir išstūmimą, nustatytą PET, ar kitus diagnostinius parametrus. Šių tyrimų subjektai gali būti pacientai iš tikslinės populiacijos arba sveiki savanoriai.
Tokiais atvejais reikalingų neklinikinių duomenų apimtis ir tipas priklausys nuo poveikio žmogui dydžio, atsižvelgiant į didžiausią klinikinę dozę ir vartojimo trukmę. Penki skirtingi tiriamųjų klinikinių tyrimų pavyzdžiai yra sugrupuoti ir išsamiau aprašyti toliau ir 3 lentelėje, įskaitant ikiklinikinių tyrimų programas, kurios gali būti rekomenduojamos tokiais atvejais. Taip pat galima naudoti alternatyvius metodus, kurie nėra aprašyti šiame standarte, įskaitant metodus, pagrindžiančius klinikinius biotechnologinių vaistų tyrimus. Alternatyvius tiriamųjų klinikinių tyrimų metodus rekomenduojama aptarti ir susitarti su atitinkamomis reguliavimo institucijomis. Bet kuris iš šių metodų gali sumažinti bendrą laboratorinių gyvūnų naudojimą kuriant vaistus.
Rekomenduojamos pradinės ir didžiausios dozės, naudojamos atliekant toksikologinius tyrimus, pateiktos 3 lentelėje. Visais atvejais PD ir farmakologinių parametrų nustatymas naudojant modelius in vivo ir/arba in vitro yra labai svarbus, kaip nurodyta 3 lentelėje ir 2 skyriuje, ir šiais duomenimis reikia pagrįsti pasirinktą dozę žmogui.
8.1 Klinikiniai tyrimai naudojant mikrodozę
Šiame skyriuje pateikti du skirtingi mikrodozės metodai yra išsamiau aprašyti 3 lentelėje.
Pirmuoju metodu bendra vaisto dozė turi būti ne didesnė kaip 100 mcg, kuri skiriama kiekvienam tiriamajam vienu metu (viena dozė) arba keliomis dozėmis. Tyrimas atliekamas siekiant ištirti tikslinių receptorių jungimąsi arba medžiagos pasiskirstymą audiniuose naudojant PET. Taip pat tokio tyrimo tikslas gali būti tirti PK su radioaktyviąja etikete arba be jos.
Antruoju metodu tiriamiesiems skiriamos 5 ar mažesnės ne didesnės kaip 100 mg dozės (iš viso 500 mikrogramų vienam tiriamajam). Tokie tyrimai atliekami su panašiais tikslais kaip ir naudojant aukščiau pateiktą metodą, tačiau esant mažiau aktyviems PET ligandams.
Kai kuriais atvejais gali būti tikslinga atlikti klinikinį tyrimą, naudojant mikrodozes ir į veną leidžiamą vaistą, skirtą vartoti per burną, turint visus ikiklinikinius toksikologinius duomenis apie geriamąjį vartojimą. Tačiau, remiantis turimais toksikologiniais duomenimis apie vaisto vartojimą per burną, kaip aprašyta 1 ir 3 lentelėse, galima apsvarstyti 3 metodą, kai buvo pasiektas priimtinas poveikio lygis, į veną leidžiamą mikrodozę. Tokiu atveju nerekomenduojama tirti intraveninio vietinio veikliosios medžiagos toleravimo, nes vartojama dozė yra labai maža (ne daugiau kaip 100 mcg). Jei į veną leidžiamame vaiste naudojamas naujas skiediklis, reikia ištirti vietinį skiediklio toleravimą.
8.2 Klinikiniai vienkartinės dozės subterapinio diapazono arba numatomo terapinio diapazono tyrimai
Taikant šį metodą (3 metodas), atliekamas vienos dozės klinikinis tyrimas, paprastai pradedant nuo subterapinių dozių, o vėliau didinant iki farmakologiškai veiksmingo arba numatomo terapinio diapazono (žr. 3 lentelę). Priimtinos didžiausios dozės nustatymas turėtų būti grindžiamas neklinikiniais duomenimis, tačiau ji gali būti dar labiau apribota, remiantis klinikiniais duomenimis, gautais vykstančio tyrimo metu. Naudojant šį metodą, galima, pavyzdžiui, nustatyti PK parametrus vartojant vaistą be radioaktyviosios etiketės, esant numatomai farmakodinamiškai veiksmingai dozei arba artimai jai. Kitas šio metodo taikymo pavyzdys yra tikslinio poveikio arba farmakologinio poveikio įvertinimas po vienkartinio vartojimo. Tyrimai taikant šį metodą nėra skirti palaikyti maksimalią toleruojamą klinikinę dozę (žr. išimtis, 1 lentelės a pastabą).
8.3 Klinikiniai tyrimai, naudojant kartotines dozes
Siekiant pagrįsti klinikinius tyrimus, kuriuose naudojamos kelios dozės, ikiklinikiniams tyrimams taikomi du skirtingi metodai (4 ir 5 metodai 3 lentelėje). Jais pagrįsti tyrimai gali pateisinti 14 dienų vaistų vartojimo terapinėmis dozėmis trukmę, siekiant įvertinti žmogaus PK ir PD parametrus, tačiau jie nėra naudojami toleruojamos maksimalios klinikinės dozės nustatymui pagrįsti.
4 metodas apima dviejų savaičių kelių dozių toksikologinį tyrimą su graužikais ir ne graužikais. Gyvūnams skiriamos dozės parenkamos remiantis daugkartine ekspozicijos doze, kai numatoma AUC esant didžiausiai klinikinei dozei.
5 metodas apima dviejų savaičių toksikologinį tyrimą su graužikais ir patvirtinamąjį toksikologijos tyrimą su ne graužikais, siekiant patvirtinti, kad NOAEL nėra toksiškas graužikams, kai jis skiriamas ne graužikams. Jei graužikams pastebimas toksinis poveikis, kai jis skiriamas ne graužikams, klinikinį vaisto vartojimą reikia atidėti, kol bus gauti duomenys iš vėlesnių neklinikinių tyrimų su tos rūšies gyvūnais (paprastai standartinis toksikologinis tyrimas, 5 skyrius).
3 lentelė. Rekomenduojami ikiklinikiniai tyrimai, pagrindžiantys galimybę atlikti tiriamuosius klinikinius tyrimus
Klinikiniai tyrimai | Ikiklinikiniai tyrimai |
|||
Skiriamos dozės | Pradinė ir maksimali dozė | Farmakologija | Bendrieji toksiškumo tyrimai | Genotoksiškumo tyrimas |
Bendra dozė 100 mikrogramų (be dozavimo intervalo), o bendra dozė yra 1/100 NOAEL ir 1/100 farmakologinės | Pradinė ir maksimali dozės gali būti vienodos, tačiau neturi viršyti bendros 100 mikrogramų dozės | Tikslo/receptoriaus profilis in vitro turi būti įvertinta | Išplėstinis vienkartinės dozės toksikologinis tyrimas (žr. c ir d pastabas) su vienos rūšies gyvūnais, paprastai graužikais, naudojant siūlomą klinikiniam naudojimui skirtą įvedimo būdą toksikokinetiniam poveikiui pasiekti. | Veiksmingoms radioaktyviosioms etiketėms (pvz., PET etiketėms) tinka |
Bendra kumuliacinė dozė yra 500 mikrogramų, ne daugiau kaip 5 vaisto dozės su išplovimo periodu tarp dozių (6 ar daugiau faktinių arba numatomų). | Pradinė ir maksimali dozės gali būti vienodos, tačiau neturi viršyti 100 mcg | Tikslo/receptoriaus profilis in vitro turi būti įvertinta Norint pagrįsti dozės, skirtos vartoti žmonėms, pasirinkimą, naudojant farmakologiškai tinkamą modelį, reikia gauti išsamius duomenis apie pagrindinius (pirminius) farmakologinius parametrus (veikimo mechanizmą ir (arba) poveikį). | Toksikologinis tyrimas, trunkantis 7 dienas, pakartotinai skiriant tos pačios rūšies gyvūnams, dažniausiai graužikams, naudojant klinikiniam naudojimui siūlomą įvedimo būdą toksikokinetikai gauti Turi būti gauti hematologiniai, klinikiniai laboratoriniai, skrodimo ir histopatologiniai duomenys Galima naudoti didžiausią dozę, 1000 kartų didesnę už klinikinę dozę, perskaičiuotą į mg/kg, kai vartojamas IV, ir mg/m2, kai vartojamas per burną. | Genotoksiškumo tyrimo atlikti nereikia, tačiau visi atlikti SAR tyrimai ar vertinimai turi būti įtraukti į klinikinio tyrimo leidimo dokumentus. Veiksmingų radioaktyviųjų atsekamųjų medžiagų (pvz., PET atsekamųjų medžiagų) atveju turėtų būti pateikti atitinkami atsekamųjų medžiagų parametrų PK įverčiai ir dozimetriniai duomenys. |
Vienos dozės subterapiniai tyrimai | Pradinė pradinė dozė turėtų būti parenkama remiantis toksikologiniais duomenimis, gautais iš jautriausių laboratorinių gyvūnų rūšių, ir duomenimis apie farmakologiškai veiksmingą dozę. Taip pat reikėtų atsižvelgti į nacionalines rekomendacijas dėl pradinės pradinės dozės žmonėms parinkimo. Didžiausia dozė gali būti ne didesnė kaip 1/2 poveikio NOAEL jautriausių rūšių laboratoriniams gyvūnams tais atvejais, kai gyvūnams pastebėtas bet koks reikšmingas toksinis poveikis yra įmanomas ir grįžtamas žmonėms. | Tikslo/receptoriaus profilis in vitro turi būti įvertinta Norint pagrįsti dozės, skirtos žmonėms, pasirinkimą, naudojant farmakologiškai reikšmingą modelį, reikia gauti išsamius duomenis apie pagrindinius (pirminius) farmakologinius parametrus (veikimo mechanizmą ir (arba) poveikį). Pagrindinis farmakologinio saugumo tyrimų rinkinys (žr. 2 skyrių) | Išplėstinis vienkartinės dozės toksikologinis tyrimas (žr. c pastabas) pagal numatytą klinikinį vaisto vartojimo būdą, gaudamas toksikokinetinius, hematologinius, laboratorinius klinikinius duomenis, skrodimo duomenis ir histopatologinį tyrimą. Šiuo atveju MTD, MFD arba ribota didelė dozė naudojama kaip didelė dozė (žr. 1.1). |
|
Vaisto vartojimas 14 dienų terapijoje | Jei toksinis poveikis pasireiškia abiejų tipų laboratoriniams gyvūnams, reikia vadovautis nacionaliniais pradinės klinikinės dozės parinkimo reikalavimais. Jei toksinis poveikis nebuvo pastebėtas jokiai laboratorinių gyvūnų rūšiai (t. y. NOAEL atitinka didžiausias ikiklinikinių tyrimų metu ištirtas dozes ir naudojamos dozės nebuvo niekaip ribojamos, pavyzdžiui, neatspindi MFD) arba buvo pastebėtas tik vienos rūšies laboratoriniams gyvūnams, tada pradinė klinikinė dozė turi būti viena iš dozių, kuriomis pasiekiamas numatomas klinikinis AUC (remiantis skirtingų rūšių PK modeliavimu arba mg/m2 konversija), kuri yra 1/50 NOAEL AUC gyvūnams ir kuriai esant mažesnė ekspozicija buvo gautas Nesant toksinio poveikio abiem rūšims, rekomenduojama naudoti didžiausią klinikinę dozę, neviršijančią 1/10 mažesnės ekspozicijos (AUC) bet kuriai rūšiai, gauta bet kuriai rūšiai naudojant didžiausią dozę. Jei toksinis poveikis pastebimas tik vienos rūšies gyvūnams, didžiausia klinikinė dozė neturi viršyti tos rūšies, kuriai buvo pastebėtas toksinis poveikis, NOAEL arba 1/2 didžiausios suleistos dozės, kuriai toksinio poveikio nepasireiškė, AUC (atsižvelgiant į tai, kuri yra žemesnė). Jei toksinis poveikis pasireiškia abiem gyvūnų rūšims, didžiausios klinikinės dozės pasirinkimas turėtų būti pagrįstas standartiniu rizikos vertinimo metodu, o šiuo ypatingu atveju galima įvertinti klinikinę MTD. | Tikslo/receptoriaus profilis in vitro turi būti įvertinta Norint pagrįsti dozės, skirtos žmonėms, pasirinkimą, naudojant farmakologiškai reikšmingą modelį, reikia gauti išsamius duomenis apie pagrindinius (pirminius) farmakologinius parametrus (veikimo mechanizmą ir (arba) poveikį). Pagrindinis farmakologinio saugumo tyrimų rinkinys (žr. 2 skyrių), naudojant dozes, panašias į bendras toksikologijos dozes | Toksikologinis tyrimas, trunkantis 14 dienų su pakartotiniu skyrimu graužikams ir negraužikams, naudojant standartinį įvertintų parametrų rinkinį; vartojama dozė parenkama atsižvelgiant į daugkartinę tikėtino klinikinio AUC ekspoziciją vartojant didžiausią dozę | Ames testas (arba alternatyvus testas, jei Ames testas yra nepriimtinas, pavyzdžiui, antibakteriniams |
Vaisto vartojimas per 14 dienų, neviršijant trukmės | Numatoma ekspozicija skiriant pradines dozes neturėtų viršyti 1/50 jautriausių gyvūnų rūšių NOAEL, skaičiuojant mg/m. Reikėtų atsižvelgti į nacionalines rekomendacijas dėl pradinės klinikinės dozės parinkimo Didžiausia ekspozicija žmonėms neturi viršyti AUC esant NOAEL ne graužikams arba 1/2 AUC esant NOAEL graužikams, atsižvelgiant į tai, kuri vertė yra mažesnė. | Tikslo/receptoriaus profilis in vitro turi būti įvertinta Norint pagrįsti dozės, skirtos žmonėms, pasirinkimą, naudojant farmakologiškai reikšmingą modelį, reikia gauti išsamius duomenis apie pagrindinius (pirminius) farmakologinius parametrus (veikimo mechanizmą ir (arba) poveikį). Pagrindinis farmakologinio saugumo tyrimų rinkinys (žr. 2 skyrių), naudojant dozes, panašias į bendrųjų toksikologijos tyrimų | Standartinis 14 dienų kartotinių dozių toksikologinis tyrimas su graužikais (su pagrindimu, kodėl graužikai atrinkti kaip priimtina laboratorinių gyvūnų rūšis šiam tyrimui). Didelė dozė yra MTD, MFD arba ribota didelė dozė (žr. 1.1). Patvirtinamasis tyrimas su ne graužikais n=3), kai numatoma NOAEL ekspozicija graužikams yra mažiausiai 3 dienos ir trumpiausia numatomo klinikinio tyrimo trukmė Norint pasiekti NOAEL poveikį graužikams, galima atlikti alternatyvų ne graužikų dozės didinimo tyrimą, kuris trunka mažiausiai 3 dienas ir trumpiausią numatomo klinikinio tyrimo trukmę. | Ames testas (arba alternatyvus testas, jei Ames testas yra nepriimtinas, pavyzdžiui, antibakteriniams |
Bendrieji ikiklinikiniai toksiškumo tyrimai turi būti atliekami pagal GLP taisykles. Genotoksiškumo tyrimo planas ir dozės parinkimas aprašyti ICH gairėse S2B. Vienos dozės išplėstinio tyrimo planas paprastai turėtų apimti hematologinių, laboratorinių, klinikinių, skrodimo ir histopatologinių duomenų įvertinimą (tik kontrolinės ir didelės dozės skiriamos, jei vartojant didelę dozę vaisto toksiškumo nepastebėta) po vienos dozės, po to stebint dvi savaites, siekiant įvertinti uždelstą toksinį poveikį ir (arba) jo išnykimą. Į standartinį graužikų tyrimo planą įtrauktas 10 gyvūnų/lyties/grupės toksikologinis įvertinimas praėjus vienai dienai po vaisto vartojimo, o 5 gyvūnai/lytis gavo pasirinktą(-as) dozę(-as), įvertintą 14 dieną po dozės suleidimo. Standartinis tyrimo be graužikų planas apima 3 gyvūnų / lyties / grupių vertinimus visoms grupėms 2 dieną po dozės suleidimo ir 2 gyvūnus / lytį, priskirtą pasirinktai (-oms) dozei (-ėms), įvertintą 14 dieną po dozės suleidimo. Vienkartinės dozės lygis toksinio poveikio grįžtamumui/uždelsimui įvertinti praėjus 14 dienų po dozės suleidimo gali būti naudojamas mikrodozės metodui pagrįsti. Dozė, naudojama gyvūnui, neturėtų būti nustatyta kaip didelė, bet turi būti bent 100 kartų didesnė už klinikinę dozę. Jei klinikinių tyrimų metu nepastebėta neigiamo poveikio, dozės didinimas virš šio AUC gali būti priimtinas, jei toksikologiniai duomenys rodo, kad galimas nepageidaujamas poveikis žmonėms yra aptinkamas, grįžtamas ir nedidelio sunkumo. |
||||
Pageidautina, kad klinikinių tyrimų metu vietinis toleravimas taikant siūlomą vartojimo būdą būtų tiriamas kaip bendro toksiškumo tyrimo dalis; Individualūs tyrimai paprastai nerekomenduojami.
Siekiant pateisinti ribotus klinikinius alternatyvaus terapinio vartojimo būdo tyrimus (pavyzdžiui, vienkartinį į veną suleidus, siekiant nustatyti absoliutų peroralinio vaisto biologinį prieinamumą), priimtinas vienos rūšies gyvūnų vienos dozės toleravimo tyrimas. Tais atvejais, kai numatoma sisteminė ekspozicija (AUC ir Cmax) neterapiniu vartojimo būdu buvo ištirta atlikus nustatytus toksikologinius tyrimus, vietinio toleravimo tyrimo baigtis gali apsiriboti klinikiniu poveikiu ir makroskopiniu bei mikroskopiniu vartojimo vietos tyrimu. Vietiniams toleravimo tyrimams skirto vaistinio preparato sudėtis gali būti ne identiška, bet turėtų būti panaši į klinikiniuose tyrimuose naudojamo vaistinio preparato sudėtį ir dozavimo formą.
Atliekant IV mikrodozės tyrimus, kurie atliekami, kai yra toksikologinių duomenų per burną (žr. 7 skyrių), farmacinės medžiagos vietinio toleravimo vertinimas nebūtinas. Jei į veną vartojamo vaisto sudėtyje naudojamas naujas tirpiklis, būtina ištirti jo vietinį toleravimą.
Jei reikia, prieš skiriant vaistinį preparatą dideliam skaičiui pacientų, reikia atlikti parenterinių vaistinių preparatų vietinio toleravimo nenumatytose injekcijos vietose tyrimus (pavyzdžiui, prieš III fazės klinikinius tyrimus). Požiūris į tokių studijų planavimą įvairiose šalyse skiriasi. Tokie tyrimai Jungtinėse Valstijose nereikalingi (išimtis būtų intratekalinis vartojimas, kai planuojama epidurinė injekcija). Japonijoje ir ES šalyse IV būdu rekomenduojama atlikti vieną paraveninę injekciją. Būtinybė ištirti kitus parenterinio vartojimo būdus vertinama individualiai.
Manoma, kad atlikti genų mutacijų tyrimus pakanka visiems vienos dozės klinikiniams tyrimams. Norint paremti kelių dozių klinikinius tyrimus, reikia atlikti papildomus tyrimus, skirtus žinduolių chromosomų pažeidimams nustatyti. Prieš pradedant II fazės klinikinius tyrimus, turi būti atlikta visa genotoksiškumo tyrimų grupė.
Jei tyrimo rezultatai rodo genotoksinio poveikio buvimą, būtina juos įvertinti ir, galbūt, atlikti papildomus tyrimus, siekiant nustatyti tolesnio vaisto vartojimo žmonėms priimtinumą.
Genotoksiškumo tyrimai, rekomenduojami siekiant paremti tiriamuosius klinikinius tyrimus taikant skirtingus metodus, yra aptarti šio standarto 8 skirsnyje.
Atvejai, kuriems reikia kancerogeniškumo tyrimų, aptariami ICH S1A gairėse dėl vaistų kancerogeniškumo tyrimų poreikio vertinimo. Tokiais atvejais kancerogeniškumo tyrimai turi būti atlikti prieš pradedant valstybinės registracijos procedūrą. Tais atvejais, kai yra įtikinamų priežasčių, rodančių kancerogeninį pavojų, tyrimų rezultatai turi būti pateikti prieš atliekant klinikinius tyrimus. Ilga klinikinio tyrimo trukmė nėra laikoma privaloma kancerogeniškumo tyrimų priežastimi.
Reikalingi vaistų, sukurtų suaugusiųjų ir vaikų sunkioms ligoms gydyti, kancerogeniškumo tyrimai gali būti atliekami, susitarus su reguliavimo institucija, po jų valstybinės registracijos.
Toksiškumo reprodukcijai tyrimai turėtų būti atliekami atsižvelgiant į pacientų populiaciją, kuri vartos tiriamąjį vaistą.
12.1 Vyrai
Patinai gali būti įtraukti į I ir II fazės klinikinius tyrimus prieš patinų reprodukcijos vertinimą dėl to, kad patinų reprodukcijos vertinimas atliekamas atliekant kartotinių dozių toksiškumo tyrimus.
2 pastaba. Patinų ir patelių vaisingumo įvertinimas atliekant standartinį sėklidžių ir kiaušidžių histologinį tyrimą atliekant mažiausiai 2 savaičių trukmės toksiškumo tyrimus (pakartotinai vartojant, dažniausiai graužikams), toksinio poveikio nustatymo gebėjimas laikomas panašiu į vaisingumo tyrimus. toksinis poveikis vyrų ir moterų reprodukciniams organams
Vyrų vaisingumo tyrimai turi būti baigti prieš pradedant plataus masto arba ilgalaikius klinikinius tyrimus (pvz., III fazės tyrimus).
12.2 Vaisingo amžiaus moterys
Jei buvo atlikti atitinkami kartotinių dozių toksiškumo tyrimai (įskaitant moterų reprodukcinių organų įvertinimą), į klinikinius tyrimus leidžiama įtraukti moteris, neturinčias reprodukcinio potencialo (t. y. nuolat sterilizuotas, po menopauzės), neatliekant toksinio poveikio reprodukcijai tyrimų. Postmenopauzė apibrėžiama kaip menstruacijų nebuvimas 12 mėnesių be kitų medicininių priežasčių.
12.3 Vaisingo amžiaus moterys
Vaisingo amžiaus moterims (WOCBP) yra didelė netyčinio vaisto poveikio embrionui ar vaisiui rizika, kol dar nėra žinoma apie galimą naudą ir riziką. Visos šalys, taikančios ICH gaires, turi panašias rekomendacijas dėl toksinio poveikio reprodukcijai tyrimų laiko, siekiant įtraukti WSDP į klinikinius tyrimus.
Įtraukiant WSDP į tyrimus, reikia nustatyti ir sumažinti netyčinio poveikio embrionui ar vaisiui riziką. Pirmasis būdas pasiekti šį tikslą yra atlikti toksinio poveikio reprodukcijai tyrimus, siekiant įvertinti vaisto keliamą riziką ir imtis atitinkamų atsargumo priemonių atliekant klinikinius tyrimus su diabetu sergančiomis moterimis. Antrasis būdas yra apriboti riziką imantis atsargumo priemonių, kad būtų išvengta nėštumo klinikinių tyrimų metu. Šios priemonės apima nėštumo testus (pvz., laisvą (3 subvienetų) hCG), labai patikimų kontracepcijos metodų naudojimą (3 pastaba) ir įtraukimą į tyrimą tik patvirtinus mėnesines. Turėtų pakakti atlikti klinikinius nėštumo testus ir pacientą informuoti. užtikrinti, kad nėštumo prevencijos priemonės būtų įgyvendinamos vaisto poveikio laikotarpiu (kuris gali viršyti tyrimo trukmę). Siekiant užtikrinti šiuos metodus, informuotas sutikimas turėtų būti pagrįstas visa turima informacija apie toksinį poveikį reprodukcijai, pavyzdžiui: galimo panašios struktūros ar farmakologinio poveikio vaistinių preparatų toksiškumo įvertinimas.Jei nėra reikšmingos informacijos apie poveikį reprodukcijai, pacientas turi būti informuojamas apie galimą nenustatytą pavojų embrionui ar vaisiui.
Visose šalyse, kurios taiko ICH gaires, esant tam tikroms sąlygoms, VSD leidžiama įtraukti į ankstyvosios fazės klinikinius tyrimus be ikiklinikinių toksiškumo vystymuisi tyrimų (pvz., neatliekant galimo poveikio embriono ir vaisiaus vystymuisi tyrimų). Vienas iš tokių reikalavimų yra, kad nėštumo rizika būtų tinkamai stebima trumpalaikių (pvz., 2 savaičių) klinikinių tyrimų metu. Kita sąlyga gali būti ligos vyravimas tarp moterų, kai neįtraukus WSDP neįmanoma pasiekti tyrimo tikslo, o pastojimo prevencijai buvo imtasi pakankamai priemonių (žr. aukščiau).
3 pastaba. Labai patikimais kontracepcijos metodais laikomi ir vienkartiniai, ir kombinuoti vaistai, kurie, nuosekliai ir teisingai vartojant, užtikrina mažą nėštumo dažnį (t. y. mažiau nei 1 % per metus). Pacientams, vartojantiems hormoninius kontraceptikus, būtina pateikti informaciją apie tiriamojo vaisto poveikį kontracepcijai.
Papildomas WSDP tyrimų atlikimo be ikiklinikinių toksinio poveikio vystymuisi tyrimų pagrindimas yra žinios apie vaisto veikimo mechanizmą, jo savybes, poveikio vaisiui trukmę arba sunkumai atliekant toksiškumo vystymuisi tyrimus su tinkamu gyvūno modeliu. Pavyzdžiui, monokloninių antikūnų, kurie, remiantis dabartiniais moksliniais duomenimis, organogenezės metu turi silpną poveikį embrionui ir vaisiui, toksiškumo vystymuisi tyrimai gali būti atliekami III fazės klinikinių tyrimų metu. Užbaigto tyrimo ataskaita turi būti pateikta kaip registracijos dokumentacijos dalis.
Apskritai, jei yra preliminarių duomenų apie toksinį poveikį reprodukcijai dviejų rūšių gyvūnams (4 pastaba) ir jei taikomos priemonės, skirtos apsisaugoti nuo nėštumo (žr. aukščiau), įtraukiant WSDP (iki 150 tiriamųjų), gaunančių tiriamąjį vaistą santykinai trumpą laikotarpį (iki 3 mėnesių), kol bus atlikti specifiniai toksiškumo reprodukcijai tyrimai. To priežastis yra labai mažas nėštumo lygis tokio dydžio ir trukmės kontroliuojamuose tyrimuose (5 pastaba) ir gerai suplanuotų bandomųjų tyrimų galimybė nustatyti svarbiausius toksiškus vystymuisi, galinčius parodyti WSDP įtraukimo į klinikinius tyrimus riziką. Moterų, įtrauktų į tyrimą, skaičiui ir tyrimo trukmei įtakos gali turėti populiacijos ypatybės, mažinančios nėštumo tikimybę (pvz., amžius, liga).
4 pastaba. Jei dozės yra tinkamos, tinkamas preliminarus embriono ir vaisiaus vystymosi tyrimas, apimantis vaisiaus išgyvenamumo, kūno masės, išorinių ir vidaus organų tyrimus, naudojant ne mažiau kaip šešias pateles grupėje, įskaitant pateles, gydomas vaistu. šiam tikslui pasiekti.organogenezės laikotarpis. Tokie preliminarūs ikiklinikiniai tyrimai turi būti atliekami laikantis aukštų mokslinių standartų, nesant lengvos prieigos prie duomenų, arba laikantis GLP reikalavimų.
5 pastaba. Moterų, kurios bando pastoti pirmą kartą, nėštumo dažnis yra maždaug 17% per menstruacinį ciklą. Nėštumo dažnis III fazės tyrimų, atliktų su vaisingo amžiaus moterimis, metu yra<0,1% на менструальный цикл. В ходе этих исследований пациентов следует предупредить о нежелательности наступления беременности и необходимости соблюдения мер по предупреждению беременности. По имеющимся данным, частота наступления беременности во II фазе ниже, чем в III фазе, но в силу ограниченного количества включенных женщин величину снижения установить невозможно. Основываясь на данных III фазы, частота наступления беременности во II фазе исследований, включающих 150 женщин с сохраненным детородным потенциалом и продолжительностью до 3 месяцев, значительно меньше 0,5 беременностей на лекарственный препарат, находящийся в разработке.
Jungtinėse Amerikos Valstijose embriono ir vaisiaus vystymosi tyrimai gali būti atidėti iki III fazės tyrimų, įtraukiant VSD, kartu imantis nėštumo prevencijos priemonių (žr. aukščiau). ES ir Japonijoje (išskyrus šiame skirsnyje aprašytas situacijas) prieš įtraukiant WSDP į tyrimą turi būti atlikti specialūs toksiškumo vystymuisi tyrimai.
Visose šalyse, įgyvendinančiose ICH gaires, WSDP leidžiama įtraukti į I ir II fazės kartotinių dozių klinikinius tyrimus prieš patelių vaisingumo tyrimus, atsižvelgiant į tai, kad gyvūnų reprodukcinės sistemos vertinimas atliekamas kaip daugkartinių dozių toksiškumo tyrimų dalis (2 pastaba). Norint įtraukti WSDP į didelio masto ilgalaikius klinikinius tyrimus (pvz., III fazės tyrimus), reikia atlikti tam skirtus ikiklinikinius moterų vaisingumo tyrimus.
Visose šalyse, kurios taiko ICH rekomendacijas, valstybinei vaistinio preparato registracijai turi būti pateikti prenatalinės ir postnatalinės otgenetinės raidos tyrimų rezultatai.
Prieš įtraukiant į bet kokius tyrimus su WHDC, kurie nenaudoja labai veiksmingų kontracepcijos metodų (3 pastaba) arba kurių nėštumo būklė nežinoma, reikia pateikti duomenis iš užbaigto toksiškumo reprodukcijai tyrimo ir standartizuotą genotoksiškumo tyrimų grupę.
12.4 Nėščios moterys
Prieš įtraukiant nėščias moteris į klinikinius tyrimus, turi būti atliktas išsamus toksiškumo reprodukcijai tyrimas ir standartizuota genotoksiškumo tyrimų grupė. Be to, būtina įvertinti turimus duomenis apie vaisto saugumą žmonėms.
Pateisinant vaikų įtraukimą į klinikinius tyrimus, svarbiausia informacija yra saugumo duomenys, gauti iš ankstesnių suaugusių pacientų tyrimų, kurie turėtų būti prieinami prieš pradedant tyrimus su vaikais. Suaugusiųjų klinikinių tyrimų duomenų pakankamumas ir apimtis šiam sprendimui pagrįsti nustatoma kiekvienu konkrečiu atveju. Prieš vartojant vaikams, gali nebūti pakankamai duomenų apie suaugusiųjų vartojimo patirtį (pavyzdžiui, esant tik vaikams skirtoms indikacijoms).
Prieš pradedant tyrimus su vaikais, reikia užpildyti tinkamos trukmės kartotinių dozių toksiškumo tyrimų su suaugusiais gyvūnais rezultatus (žr. 1 lentelę), pagrindinį farmakologinio saugumo tyrimų rinkinį ir standartinį genotoksiškumo tyrimų rinkinį. Taip pat gali prireikti tirtų vaikų amžių ir lytį atitinkančių toksiškumo reprodukcijai duomenų, kuriuose pateikiama informacija apie tiesioginį toksinį pavojų arba poveikį vystymuisi (pvz., vaisingumo tyrimai, prenatalinis ir postnatalinis vystymasis). Embriono ir vaisiaus vystymosi tyrimai nėra labai svarbūs siekiant pagrįsti klinikinius tyrimus su vyrais arba prieš brendimą sulaukusiomis moterimis.
Bet kokių tyrimų su nesubrendusiais gyvūnais būtinybė turėtų būti svarstoma tik tuo atveju, jei manoma, kad ankstesni gyvūnų ir žmonių saugumo duomenys, įskaitant kitų tos pačios farmakologinės klasės vaistų poveikį, yra nepakankami klinikiniam tyrimui su vaikais pagrįsti. Jei tokie ikiklinikiniai tyrimai būtini, pakanka naudoti vieną gyvūnų rūšį, geriausia – graužikus. Jei yra pakankamai mokslinio pagrindimo, leidžiama atlikti ne graužikų tyrimus.
Atliekant trumpalaikius PK tyrimus su vaikais (pvz., 1–3 dozėmis), toksiškumo tyrimai su nesubrendusiais gyvūnais paprastai nėra laikomi informatyviais.
Atsižvelgiant į vartojimo indikacijas, į klinikinį tyrimą įtrauktų vaikų amžių ir suaugusių gyvūnų bei pacientų saugumo duomenis, prieš pradedant trumpalaikius klinikinius veiksmingumo tyrimus reikia atsižvelgti į poreikį gauti tyrimų su nesubrendusiais gyvūnais rezultatus. naudojant platų dozių diapazoną ir vaisto saugumą.vaistas. Vienas iš svarbiausių klausimų yra tyrimo dalyvių amžius, susijęs su tyrimo trukme (tai yra vystymosi laikotarpio, per kurį tyrimo dalyviai vartoja vaistą, dalis). Šis veiksnys yra lemiamas vertinant poreikį atlikti ikiklinikinius tyrimus su nesubrendusiais gyvūnais ir, jei jie būtini, turėtų būti nustatytas jų atlikimo laikas, susijęs su klinikiniais tyrimais.
Prieš pradedant ilgalaikius klinikinius tyrimus su vaikais, kuriems reikia toksiškumo tyrimų su nesubrendusiais gyvūnais, šie ikiklinikiniai tyrimai turi būti baigti.
Gali būti situacijų, kai vaikai yra pagrindinė gydomoji populiacija, o turimi eksperimentiniai duomenys rodo galimą tiriamojo vaisto poveikį organų taikinių vystymuisi (toksikologinį ar farmakologinį). Kai kuriais iš šių atvejų gali prireikti ilgalaikių tyrimų su nesubrendusiais gyvūnais. Ilgalaikis toksikologinis tyrimas su atitinkamos rūšies ir amžiaus gyvūnais yra priimtinas (pvz., 12 mėnesių trukmės tyrimas su šunimis arba 6 mėnesių tyrimas su žiurkėmis). 12 mėnesių trukmės tyrimas gali apimti visą šunų vystymosi laikotarpį. Kitoms laboratorinių gyvūnų rūšims ši konstrukcija gali būti pritaikyta pakeisti atitinkamą standartinį lėtinį tyrimą ir atskirą tyrimą su nesubrendusiais gyvūnais tam tikromis sąlygomis.
Prieš pradedant ilgalaikius klinikinius tyrimus su vaikais, būtina nustatyti kancerogeniškumo tyrimų poreikį. Tačiau, jei nėra svarbių priežasčių (pavyzdžiui, įvairių bandymų metu pateikti toksinio poveikio kepenims įrodymai arba prokancerogeninė rizika dėl veikimo mechanizmo ar poveikio, nustatyto bendrojo toksiškumo tyrime), kancerogeniškumo tirti nereikia. klinikiniams tyrimams su vaikais.
Kaip nurodyta ICH S8 rekomendacijose dėl vaistų imunotoksiškumo tyrimų, visi nauji vaistai turėtų būti įvertinti dėl imunotoksinio poveikio naudojant standartinius toksikologijos tyrimus ir papildomus imunotoksiškumo tyrimus, atliktus remiantis įrodymų visuma, įskaitant imuninės sistemos sukeltus signalus, nustatytus atliekant standartinius toksikologijos tyrimus. . Jei reikia atlikti papildomus imunotoksiškumo tyrimus, juos reikia atlikti prieš naudojant tiriamąjį vaistą didesnėms pacientų grupėms (pvz., III fazės klinikiniai tyrimai).
Fotosaugos tyrimo poreikis arba laikas, atsižvelgiant į žmogaus apšvitą, nustatomi:
- fotocheminės molekulės savybės (pavyzdžiui, fotoabsorbcija ir fotostabilumas);
- informacija apie chemiškai panašių junginių fototoksinį potencialą;
- pasiskirstymas audiniuose;
- klinikiniai arba ikiklinikiniai duomenys, rodantys fototoksiškumo buvimą.
Pradinis fototoksinio potencialo įvertinimas turi būti atliktas remiantis fotocheminėmis vaisto savybėmis ir jo farmakologine/chemine klase. Jei įvertinus visus turimus duomenis ir siūlomą klinikinio tyrimo planą, paaiškėja, kad žmonėms kyla didelė fototoksiškumo rizika, ambulatorinių klinikinių tyrimų metu turi būti taikomos pacientų apsaugos priemonės. Be to, norint gauti informacijos apie pavojų žmonėms ir tolesnio tyrimo poreikį, būtinas tolesnis veikliosios medžiagos pasiskirstymo odoje ir akyse įvertinimas. Tada, jei taikoma, eksperimentinis įvertinimas (ikiklinikinis, in vitro arba in vivo, arba klinikinis) fototoksiškumo potencialas turi būti atliktas prieš vartojant vaistą daugeliui pacientų (III fazės klinikiniai tyrimai).
Arba, vietoj pirmiau aprašyto laipsniško metodo, tiesioginis fototoksinio potencialo įvertinimas gali būti atliktas atliekant ikiklinikinius arba klinikinius tyrimus. Jei šių tyrimų rezultatai yra neigiami, klinikinio tyrimo metu nereikia anksti įvertinti vaistų pasiskirstymo akyse/odoje ir imtis prevencinių priemonių.
Jei fototoksiškumo vertinimo rezultatai rodo galimą fotokancerogeninį potencialą, rizika paprastai yra tinkamai kontroliuojama pacientams taikant apsaugos priemones, įskaitant įspėjimą informuotame sutikime ir naudojimo instrukciją (žr. 6 pastabą).
6 pastaba. Fotokancerogeniškumo tyrimas ne graužikams naudojant šiuo metu turimus modelius (pvz., beplaukius graužikus) vaistams kurti yra laikomas nepraktišku ir paprastai nėra būtinas. Jei fototoksiškumo tyrimai rodo galimą fotokancerogeniškumo riziką ir atsiranda tinkamas tyrimo metodas, tyrimą paprastai reikia užbaigti prieš pradedant registracijos procesą ir į rezultatus turi būti atsižvelgta vertinant riziką žmonėms.
Vaistams, kurie veikia centrinę nervų sistemą, neatsižvelgiant į vartojimo indikacijas, būtina nustatyti, ar reikia įvertinti priklausomybės nuo narkotikų išsivystymo riziką. Ikiklinikiniai tyrimai būtini norint pagrįsti klinikinių tyrimų planą, nustatyti specialią šalyje naudojamą kategoriją (pavyzdžiui, narkotinių ir psichotropinių medžiagų sąrašus ir kt.), parengti vartojimo instrukcijas. Sudarant būtinų tyrimų rinkinį, reikia vadovautis nacionalinėmis gairėmis dėl ikiklinikinio vertinimo ir priklausomybės nuo narkotikų išsivystymo rizikos.
Ikiklinikiniai duomenys, surinkti vaistų kūrimo pradžioje, gali būti informatyvūs nustatant ankstyvus priklausomybės potencialo rodiklius. Duomenys apie tokius ankstyvus rodiklius turėtų būti gauti prieš pirmą kartą vartojant vaistą žmonėms; Tai apima PK / PD profilį, kad būtų galima nustatyti veikimo trukmę, cheminės struktūros panašumą į piktnaudžiavimo vaistus, receptorių jungimosi profilį ir elgesio / klinikinius simptomus iš ikiklinikinių tyrimų. in vivo. Jei šie ankstyvieji tyrimai neatskleidžia priklausomybės nuo narkotikų galimybės, gali prireikti atlikti išsamių ikiklinikinių priklausomybės nuo narkotikų modelių tyrimų. Apskritai, jei veiklioji medžiaga pasižymi ypatumais, panašiais į žinomus priklausomybės nuo vaistų modelius arba turi naują veikimo mechanizmą centrinei nervų sistemai, prieš pradedant didelius klinikinius tyrimus (pvz., III fazės klinikinius tyrimus), rekomenduojama atlikti papildomus ikiklinikinius tyrimus.
Jei metabolitų profilis ir vaisto veikimo tikslas graužikams sutampa su žmonėmis, graužikams atliekamas neklinikinis priklausomybės nuo vaistų rizikos vertinimas. Nežmoginiai primatai turėtų būti naudojami tik retais atvejais, kai yra įtikinamų įrodymų, kad tokie tyrimai numato žmonių jautrumą priklausomybei nuo narkotikų, o graužikų modeliai yra netinkami. Priklausomybės nuo narkotikų išsivystymo rizikai įvertinti dažniausiai atliekami trijų tipų tyrimai: pirmenybės vaistams, vaistų savarankiško vartojimo ir įvertinimas po vaisto vartojimo nutraukimo. Vaistų pasirinkimo ir savarankiško vartojimo tyrimai paprastai atliekami kaip atskiri eksperimentai. Nutraukimo tyrimai kartais gali būti įtraukti į kartotinės dozės toksiškumo tyrimą (toksiškumo grįžtamumo skydelis). Manoma, kad tokiam neklinikiniam priklausomybės nuo vaistų rizikos vertinimui tinka didžiausia dozė, kurios koncentracija laboratorinių gyvūnų kraujo plazmoje yra kelis kartus didesnė už terapinę klinikinę dozę žmonėms.
Jei ankstesni ikiklinikiniai ar klinikiniai vaisto ar susijusių vaistų duomenys rodo, kad gali kilti specifinių saugumo problemų, gali prireikti papildomų ikiklinikinių tyrimų (pvz., siekiant nustatyti galimus biomarkerius, išsiaiškinti veikimo mechanizmą).
ICH gairėse Q3A ir Q3B pateikiami metodai, kaip kvalifikuoti veikliosios medžiagos priemaišas ir skilimo produktus. Jei priemaišoms ir skilimo produktams nustatyti reikalingi specialūs tyrimai, jų paprastai nereikia atlikti iki III fazės klinikinių tyrimų pradžios, nebent dėl vystymosi pokyčių atsiranda iš esmės naujas priemaišų profilis (pvz., nauji sintezės būdai). Vaisto komponentų sąveikos rezultatas). Tokiais atvejais gali prireikti atitinkamų priemaišų ir skilimo produktų kvalifikacinių tyrimų, kad būtų galima paremti II fazės ar vėlesnių vystymosi klinikinių tyrimų rezultatus.
Šis skyrius taikomas kombinuotiems vaistams, kurie yra skirti vartoti vienu metu ir yra sudėti į vieną pakuotę arba skirti vartoti vienoje dozavimo formoje („fiksuotas derinys“). Žemiau išdėstyti principai gali būti taikomi ir nekombinuotiems vaistams, kurie pagal vartojimo instrukcijas gali būti vartojami kartu su tam tikru vaistu, įskaitant ir ne „fiksuoto derinio“ pavidalu, taip pat vaistams, kuriems nepakanka klinikinių duomenų apie derinį.
Šis standartas taikomas šiems deriniams:
1) dvi ar daugiau medžiagų vėlyvose kūrimo stadijose (junginiai, turintys didelę klinikinio naudojimo patirtį (t. y. III fazės klinikiniai tyrimai arba tyrimai po registracijos);
2) viena ar kelios medžiagos vėlyvose vystymosi stadijose ir viena ar kelios medžiagos ankstyvosiose vystymosi stadijose (klinikinė patirtis yra ribota, pvz., II fazės klinikinis tyrimas ir ankstesnių fazių tyrimai), arba
3) daugiau nei viena medžiaga ankstyvosiose vystymosi stadijose.
Daugumos derinių, kurių sudėtyje yra dvi cheminės medžiagos, kurios yra vėlyvose kūrimo stadijose, bet nėra didelės klinikinės bendro naudojimo patirties, derinio toksikologiniai tyrimai nėra būtini klinikiniams tyrimams ar teisės aktų patvirtinimui pagrįsti, nebent būtų pagrindo įtarti galimą bendrą toksikologinį poveikį. (pvz., buvimas tikslinių organų, identiškų toksiniam poveikiui). Šios priežastys gali skirtis priklausomai nuo saugos lygio ir galimybės stebėti neigiamą poveikį žmonėms. Jei reikalingas neklinikinis tyrimas siekiant įvertinti galimą papildomą toksikologinį derinio poveikį, jis turi būti baigtas prieš pradedant klinikinius derinio tyrimus.
Derinių, kurių sudėtyje yra dviejų medžiagų, kurios yra vėlyvose vystymosi stadijose, tačiau nėra priimtinos klinikinės patirties kartu vartojant, derinio ikiklinikinių tyrimų, kurie paremtų santykinai trumpalaikius klinikinius tyrimus, paprastai nėra (pvz., II fazės tyrimai iki 3 mėnesiai) reikalaujama, jei remiantis pakankamais turimais duomenimis pagrįsta nuomonė, kad nėra galimo toksikologinio derinio poveikio. Tačiau ilgalaikiams ir didelės apimties klinikiniams tyrimams, taip pat valstybinės registracijos procedūrai, tokių derinių ikiklinikiniai tyrimai yra privalomi.
Ankstyvosiose kūrimo stadijose esantiems cheminių medžiagų deriniams, turintiems klinikinę patirtį ir vėlesniuose vystymosi etapuose esančiomis medžiagomis, dėl kurių nėra didelių toksikologinių problemų, derinio toksikologinių tyrimų nereikia, kad būtų galima pagrįsti „koncepcijos įrodymo“ tyrimų pagrįstumą. 1 mėnesio trukmė.. Klinikinių derinio tyrimų trukmė neturi viršyti klinikinės patirties su atskirais komponentais. Vėlesniems ir ilgesniems klinikiniams tyrimams reikalingi ikiklinikiniai derinio tyrimai.
Deriniams, kurių sudėtyje yra ankstyvosiose vystymosi stadijose esančių medžiagų, būtina atlikti ikiklinikinius jų derinio tyrimus, kad būtų galima pagrįsti klinikinių tyrimų atlikimo galimybę.
Jei buvo atlikta visa ikiklinikinių tyrimų programa kiekvienam derinio komponentui, o ikiklinikiniai derinio toksikologiniai tyrimai būtini klinikinio tyrimo atlikimo pagrįstumui pagrįsti, derinio tyrimo trukmė turėtų būti lygiavertė klinikinio tyrimo metu (bet ne ilgiau kaip 90 dienų). Šis ikiklinikinis tyrimas taip pat bus tinkamas valstybinės registracijos procedūrai. Ikiklinikiniam trumpesnės trukmės derinio tyrimui taip pat gali būti suteiktas teisės aktų patvirtinimas, atsižvelgiant į numatomo klinikinio naudojimo trukmę.
Neklinikinių tyrimų, rekomenduojamų tirti derinį, planas priklauso nuo atskirų komponentų farmakologinių, toksikologinių ir farmakokinetinių savybių, vartojimo indikacijų, siūlomos tikslinės pacientų populiacijos ir turimų klinikinių duomenų.
Ikiklinikiniai derinio tyrimai paprastai atliekami su viena tinkama gyvūnų rūšimi. Jei nustatomas netikėtas toksinis poveikis, gali prireikti papildomų tyrimų.
Tais atvejais, kai nebaigta atskirų komponentų visa neklinikinių bandymų programa, visa neklinikinės toksikologijos programa gali būti vykdoma tik deriniui, jei atskiri komponentai yra skirti naudoti tik derinyje.
Jei atskiros sudedamosios dalys buvo ištirtos pagal galiojančius standartus, klinikiniams tyrimams ar valstybinės registracijos procedūroms derinio genotoksiškumo, farmakologinio saugumo ir kancerogeniškumo tyrimai paprastai nereikalingi. Tais atvejais, kai pacientų populiacija apima WSDP, o atskiro (-ių) komponento (-ų) tyrimai rodo riziką embrionui ir vaisiui, kombinuotų tyrimų nerekomenduojama, nes jau buvo nustatyta galimybė pakenkti žmogaus embriono ir vaisiaus vystymuisi. Jei ikiklinikiniai embriono ir vaisiaus vystymosi tyrimai rodo, kad nė vienas komponentas nekelia pavojaus žmogaus vystymuisi, derinio tyrimų atlikti nereikia, nebent, remiantis atskirų komponentų savybėmis, kyla susirūpinimas, kad derinys gali kelti pavojų žmonių saugumui. Tais atvejais, kai buvo tiriamas atskirų kompozicijos komponentų poveikis embriono ir vaisiaus vystymuisi, tačiau reikalingi deriniai, pastarųjų rezultatai turi būti pateikti valstybinės registracijos procedūros pradžioje.
Santrumpos
Plotas po kreive | Plotas po farmakokinetine kreive |
|
Didžiausia plazmos koncentracija | Didžiausia koncentracija plazmoje |
|
Europos Sąjunga |
||
Gera laboratorinė praktika | Gera laboratorinė praktika |
|
Žmogaus chorioninis gonadotropinas | Žmogaus chorioninis gonadotropinas |
|
Žmogaus imunodeficito virusas | Imunodeficito virusas |
|
Tarptautinė žmonėms skirtų vaistų registravimo techninių reikalavimų derinimo konferencija | Tarptautinė medicinos reikmėms skirtų vaistų registravimo techninių reikalavimų derinimo konferencija |
|
Intraveninis |
||
Didžiausia įmanoma dozė | Didžiausia leistina dozė |
|
Didžiausia toleruojama dozė | Didžiausia toleruojama dozė |
|
VNTD (NOAEL) | Nėra pastebėto neigiamo poveikio lygio | Didelė netoksiška dozė |
Pozitronų emisijos tomografija | Pozitronų emisijos tomografija |
|
Farmakokinetika | Farmakokinetika |
|
Farmakodinamika | Farmakodinamika |
|
Struktūros ir veiklos santykis | Molekulinės struktūros aktyvumo sąlygoti santykiai |
|
Maža trukdanti RNR | Maža trukdanti RNR |
|
WSDP (WOCBP) | Moterys, turinčios vaisingumą | Vaisingo amžiaus moterys |
ICH S6 gairės: ikiklinikinis biotechnologiniais preparatais pagamintų vaistų saugos vertinimas; 1997 m. liepos mėn.
ICH E8 gairės: bendrosios klinikinių tyrimų nuostatos; 1997 m. liepos mėn.
ICH S5(R2) gairės: Vaistų toksiškumo reprodukcijai ir toksiškumo vyrų vaisingumui nustatymas; 1993 metų birželis.
ICH S1 C(R2) gairės: Dozės parinkimas farmacijos produktų kancerogeniškumo tyrimams; 2008 m. kovo mėn.
ICH S7A gairės: Farmakologijos žmonėms skirtų vaistų saugos tyrimai; 2000 metų lapkritis.
ICH S7B gairės: Neklinikinis uždelstos skilvelių repoliarizacijos (QT intervalo pailgėjimo) potencialo įvertinimas pagal žmogaus vaistus; 2005 m. gegužės mėn.
ICH S3A gairės: Toksikokinetikos rekomendacijų pastaba: sisteminio poveikio vertinimas atliekant toksiškumo tyrimus; 1994 metų spalis.
Nacionalinis tyrimų metu atliekamų gyvūnų pakeitimo, tobulinimo ir mažinimo centras. Sudėtingi reikalavimai ūmaus toksiškumo tyrimams: seminaro ataskaita; 2007 m. gegužės mėn.
Robinson S., Delongeas JL., Donald E., Dreher D., Festag M., Kervyn S. ir kt. Europos farmacijos įmonės iniciatyva, kuria ginčijamas reguliavimo reikalavimas dėl ūmaus toksiškumo tyrimų kuriant farmacinius vaistus. Regul Toxicol Pharmacol 2008;50:345-352.
ICH S2B gairės: Genotoksiškumas: standartinė baterija, skirta farmacijos produktų genotoksiškumo testavimui; 1997 m. liepos mėn.
ICH S1A gairės: Vaistų kancerogeniškumo tyrimų poreikio gairės; 1995 metų lapkritis.
ICH Q3A(R2) gairės: priemaišos naujose vaistinėse medžiagose; 2006 m. spalio mėn.
ICH Q3B(R2) gairės: priemaišos naujuose vaistų produktuose; 2006 m. birželis.
ICH S8 gairės: Žmonėms skirtų vaistų imunotoksiškumo tyrimai; 2005 m. rugsėjo mėn.
Sakai T., Takahashi M., Mitsumori K., Yasuhara K., Kawashima K., Mayahara H. ir kt. Bendras darbas, siekiant įvertinti toksiškumą patinų reprodukciniams organams atliekant 2 savaičių kartotinių dozių toksiškumo tyrimus su žiurkėmis. Studijų apžvalga. J Toxicol Sci 2000;25:1-21.
Sanbuissho A., Yoshida M., Hisada S., Sagami F., Kudo S., Kumazawa T. ir kt. Bendras darbas vertinant toksinį poveikį kiaušidėms atliekant kartotinių dozių ir vaisingumo tyrimus su žiurkių patelėmis. J Toxicol Sci 2009;34:1-22.
Paraiška TAIP
(informatyvus)
DA.1 lentelė
Nurodyto tarptautinio dokumento pavadinimas | Atitikties laipsnis | Atitinkamo nacionalinio standarto pavadinimas ir pavadinimas |
ICH S3A gairės | GOST R 56702-2015 "Medicininiai vaistai. Ikiklinikiniai toksikologiniai ir farmakokinetiniai saugumo tyrimai" |
|
ICH S6 gairės "Žmonėms skirti vaistiniai preparatai. Neklinikiniai farmakologinio saugumo tyrimai" |
||
EBPO geros laboratorinės praktikos principai | GOST R 53434-2009 „Geros laboratorinės praktikos principai“ |
|
Pastaba – šioje lentelėje naudojami šie atitikties standartams laipsnio susitarimai: IDT – identiški standartai; MOD – modifikuoti standartai. |
||
UDC 615.038:615.012/.014:615.2:006.354 | |
Raktažodžiai: vaistai, ikiklinikiniai saugumo tyrimai, klinikiniai tyrimai, valstybinė registracija, sauga |
|
Elektroninio dokumento tekstas
parengė Kodeks JSC ir patikrino, ar:
oficialus leidinys
M.: Standartinform, 2016 m
Klinikinis tyrimas (KT) – Tai tiriamo vaisto klinikinių, farmakologinių, farmakodinaminių savybių tyrimas žmonėms, įskaitant absorbcijos, pasiskirstymo, keitimo ir išskyrimo procesus, siekiant, naudojant mokslinius metodus, gauti veiksmingumo ir saugumo įvertinimus ir įrodymus. vaistų, duomenų apie numatomą šalutinį poveikį ir sąveikos su kitais vaistais poveikį.
Vaistų klinikinių tyrimų tikslas yra moksliniais metodais, vertinimais ir vaistų veiksmingumo bei saugumo įrodymais gauti duomenis apie galimą šalutinį poveikį vartojant vaistus ir sąveikos su kitais vaistais poveikį.
Atliekant klinikinius naujų farmakologinių medžiagų tyrimus, 4 tarpusavyje sujungtos fazės:
1. Nustatyti vaistų saugumą ir nustatyti toleruojamų dozių diapazoną. Tyrimas atliekamas su sveikais vyrais savanoriais, išskirtiniais atvejais – su pacientais.
2. Nustatyti vaistų veiksmingumą ir toleravimą. Parenkama mažiausia veiksminga dozė, nustatomas terapinio poveikio plotis ir palaikomoji dozė. Tyrimas atliekamas su nozologijos pacientais, kuriems yra skirtas tiriamas vaistas (50-300 asmenų).
3. Išsiaiškinamas vaisto veiksmingumas ir saugumas, jo sąveika su kitais vaistais, lyginant su standartiniais gydymo metodais. Tyrimas atliekamas su daugybe pacientų (tūkstančiai pacientų), dalyvaujant specialioms pacientų grupėms.
4. Poregistraciniais rinkodaros tyrimais tiriamas toksinis vaisto poveikis ilgalaikio vartojimo metu ir nustatomas retas šalutinis poveikis. Į tyrimą gali būti įtrauktos skirtingos pacientų grupės – pagal amžių, pagal naujas indikacijas.
Klinikinių tyrimų tipai:
Atviras, kai visi tyrimo dalyviai žino, kokį vaistą pacientas vartoja;
Paprastas „aklas“ – pacientas nežino, bet tyrėjas žino, koks gydymas buvo paskirtas;
Dvigubai aklo tyrimo metu nei tyrimo personalas, nei pacientas nežino, ar jis gauna vaistą, ar placebą;
Trigubai aklas – nei tyrėjas, nei egzaminuotojas, nei pacientas nežino, kokiu vaistu gydoma.
Viena iš klinikinių tyrimų rūšių yra bioekvivalentiškumo tyrimai. Tai yra pagrindinis generinių vaistų, kurie dozavimo forma ir veikliųjų medžiagų kiekiu nesiskiria nuo atitinkamų originalų, kontrolės būdas. Bioekvivalentiškumo tyrimai leidžia mums padaryti pagrįstą
išvados apie lyginamų vaistų kokybę remiantis mažesniu pirminės informacijos kiekiu ir per trumpesnį laiką. Jie daugiausia atliekami sveikiems savanoriams.
Visų fazių klinikiniai tyrimai atliekami Rusijoje. Didžioji dalis tarptautinių užsienio vaistų klinikinių tyrimų ir studijų priklauso 3 fazei, o vidaus vaistų klinikinių tyrimų atveju nemaža jų dalis yra 4 fazės tyrimai.
Rusijoje per pastaruosius dešimt metų specializuota klinikinių tyrimų rinka.Čia dirba gerai struktūrizuoti, aukštos kvalifikacijos specialistai – gydytojai tyrėjai, mokslininkai, organizatoriai, vadovai ir kt., aktyvios įmonės, kurios savo verslą remia organizaciniais, aptarnavimo, analitiniais klinikinių tyrimų atlikimo aspektais, tarp jų – sutartinės mokslinių tyrimų organizacijos, medicinos centrų statistika.
Nuo 1998 m. spalio mėn. iki 2005 m. sausio 1 d. patvirtinti buvo pateikta 1 840 klinikinių tyrimų. 1998-1999 metais šalies įmonės sudarė itin nedidelę besikreipiančiųjų dalį, tačiau nuo 2000 metų jų vaidmuo pastebimai išaugo: 2001 metais jų buvo 42%, 2002 metais – jau 63%, 2003 metais – 45,5%. Iš užsienio šalių kandidačių pirmenybę teikia Šveicarija, JAV, Belgija ir JK.
Klinikinių tyrimų objektai – tiek vietinės, tiek užsienio kilmės vaistai, kurių apimtys liečia beveik visas žinomas medicinos sritis. Daugiausia vaistų naudojama širdies ir kraujagyslių ligoms bei vėžiui gydyti. Po to seka tokios sritys kaip psichiatrija ir neurologija, gastroenterologija, infekcinės ligos.
Viena iš klinikinių tyrimų sektoriaus plėtros tendencijų mūsų šalyje turėtų būti spartus klinikinių tyrimų dėl generinių vaistų bioekvivalentiškumo augimas. Akivaizdu, kad tai visiškai atitinka Rusijos farmacijos rinkos ypatybes: kaip žinoma, tai generinių vaistų rinka.
Klinikinių tyrimų vykdymas Rusijoje yra reglamentuotasRusijos Federacijos Konstitucija, kuriame teigiama, kad „... niekas
gali būti atliekami medicininiai, moksliniai ir kiti eksperimentai be savanoriško sutikimo.
Kai kurie straipsniai Federalinis įstatymas „Rusijos Federacijos įstatymų dėl piliečių sveikatos apsaugos pagrindai“(1993 m. liepos 22 d., Nr. 5487-1) apibrėžia klinikinių tyrimų atlikimo pagrindus. Taigi, 43 straipsnyje nurodyta, kad vaistai, kurie nėra patvirtinti vartoti, bet yra nustatyta tvarka peržiūrimi, gali būti naudojami paciento išgydymui tik gavus jo savanorišką rašytinį sutikimą.
Federalinis įstatymas „Dėl vaistų“ 86-FZ turi atskirą IX skyrių „Vaistų kūrimas, ikiklinikiniai ir klinikiniai tyrimai“ (37-41 straipsniai). Čia nurodoma sprendimo dėl vaistų klinikinio tyrimo atlikimo priėmimo tvarka, klinikinių tyrimų atlikimo teisinis pagrindas ir klinikinių tyrimų finansavimo klausimai, jų atlikimo tvarka, klinikiniuose tyrimuose dalyvaujančių pacientų teisės.
Klinikiniai tyrimai atliekami pagal pramonės standartą OST 42-511-99 „Kokybiškų klinikinių tyrimų Rusijos Federacijoje atlikimo taisyklės“(patvirtinta Rusijos sveikatos apsaugos ministerijos 1998 m. gruodžio 29 d.) (Geroji klinikinė praktika – GCP). Kokybiškų klinikinių tyrimų atlikimo Rusijos Federacijoje taisyklės numato etinį ir mokslinį žmogaus tyrimų planavimo ir atlikimo kokybės standartą, jų rezultatų dokumentavimą ir pateikimą. Šių taisyklių laikymasis yra klinikinių tyrimų rezultatų patikimumo, saugumo, tiriamųjų teisių ir sveikatos apsaugos garantija pagal esminius Helsinkio deklaracijos principus. Atliekant klinikinius vaistinių preparatų tyrimus, kurių rezultatus planuojama pateikti licencijas išduodančioms institucijoms, būtina laikytis šių Taisyklių reikalavimų.
GCP nustato klinikinių tyrimų planavimo, vykdymo, dokumentavimo ir kontrolės reikalavimus, skirtus juose dalyvaujančių asmenų teisių, saugos ir sveikatos apsaugai užtikrinti, kurių metu negalima atmesti nepageidaujamo poveikio žmonių saugai ir sveikatai, taip pat užtikrinti gautų duomenų patikimumą ir tikslumą.informacijos tyrimo metu. Taisyklės yra privalomos visiems klinikinių vaistų tyrimų Rusijos Federacijos teritorijoje dalyviams.
Siekiant pagerinti vaistų, kurie yra pagrindinė generinių vaistų medicininės ir biologinės kontrolės rūšis, bioekvivalentiškumo tyrimų atlikimo metodinę bazę, Rusijos Federacijos sveikatos ir socialinės plėtros ministerija 2004 m. rugpjūčio 10 d. patvirtino metodines rekomendacijas. „Aukštos kokybės klinikinių vaistų bioekvivalentiškumo tyrimų atlikimas“.
Pagal norminius dokumentus, Vykdomi CI federalinės vykdomosios institucijos akredituotose sveikatos priežiūros įstaigose, kurių kompetencijai priklauso valstybinės kontrolės ir priežiūros vaistų apyvartos srityje įgyvendinimas; taip pat sudaro ir skelbia sveikatos priežiūros įstaigų, turinčių teisę atlikti klinikinius vaistų tyrimus, sąrašą.
Teisinis pagrindas atlikti klinikinius vaistų tyrimus yra federalinės vykdomosios institucijos, kurios kompetencijai priklauso valstybinės kontrolės ir priežiūros vaistų apyvartos srityje įgyvendinimas, sprendimas dėl vaisto klinikinio tyrimo atlikimo ir susitarimo dėl jo atlikimo. Sprendimą atlikti klinikinį vaistų tyrimą priima Rusijos Federacijos federalinė sveikatos priežiūros ir socialinės raidos priežiūros tarnyba, vadovaudamasi Vaistų įstatymu ir remdamasi prašymu, teigiama etikos komiteto išvada. prie federalinės vaistų kokybės kontrolės institucijos ikiklinikinių tyrimų ataskaita ir išvada bei vaisto medicininio vartojimo instrukcijos.
Prie federalinės narkotikų kokybės kontrolės institucijos buvo įkurtas Etikos komitetas. Sveikatos priežiūros įstaiga nepradeda tyrimo, kol Etikos komitetas nepatvirtina (raštu) rašytinės informuoto sutikimo formos ir kitos tiriamajam ar jo teisėtam atstovui pateiktos medžiagos. Informuoto sutikimo forma ir kita medžiaga tyrimo metu gali būti peržiūrimi, jei nustatomos aplinkybės, galinčios turėti įtakos tiriamojo sutikimui. Aukščiau išvardytos dokumentacijos nauja redakcija turi būti patvirtinta Etikos komiteto, o jos pristatymo tiriamajam faktas turi būti patvirtintas dokumentais.
Pirmą kartą pasaulio praktikoje Prūsijoje buvo sukurta ir įgyvendinta valstybinė klinikinių tyrimų vykdymo ir eksperimento dalyvių teisių laikymosi kontrolė. 1900 m. spalio 29 d. Sveikatos apsaugos ministerija įpareigojo universitetų klinikas atlikti klinikinius eksperimentus, gavus išankstinį raštišką pacientų sutikimą. 1930-aisiais Padėtis pasaulyje labai pasikeitė žmogaus teisių srityje. Karo belaisvių koncentracijos stovyklose Vokietijoje ir Japonijoje buvo atliekami tokio didelio masto žmonių eksperimentai, kad laikui bėgant kiekviena koncentracijos stovykla netgi sukūrė savo medicininių eksperimentų „specializaciją“. Tik 1947 m. Tarptautinis karinis tribunolas grįžo prie klinikiniuose tyrimuose dalyvaujančių žmonių teisių apsaugos problemos. Jos darbo metu buvo sukurtas pirmasis tarptautinis kodeksas „Eksperimentų su žmonėmis taisyklių kodeksas“ vadinamasis Niurnbergo kodeksas.
1949 m. Londone buvo priimtas Tarptautinis medicinos etikos kodeksas, skelbiantis tezę, kad „gydytojas turi veikti tik paciento interesais, teikdamas medicininę pagalbą, kuri turėtų pagerinti paciento fizinę ir psichinę būklę“, ir Ženevos tarptautinis medicinos institutas. Pasaulio gydytojų asociacijos konvencija (1948–1949) apibrėžė gydytojo pareigą žodžiais: „Mano pirmoji užduotis yra rūpintis savo paciento sveikata“.
Klinikinių tyrimų etinės sistemos kūrimo lūžis buvo 1964 m. birželio mėn. Helsinkyje vykusi 18-oji Pasaulio gydytojų asociacijos generalinė asamblėja. Helsinkio deklaracija Pasaulio medicinos asociacija, kuri perėmė visą pasaulio patirtį biomedicininių tyrimų etinio turinio srityje. Nuo tada deklaracija buvo keletą kartų peržiūrėta, paskutinį kartą Edinburge (Škotija) 2000 m. spalį.
Helsinkio deklaracijoje teigiama, kad biomedicininiai tyrimai, kuriuose dalyvauja žmonės, turi atitikti visuotinai pripažintus mokslinius principus ir būti pagrįsti tinkamai atliktais laboratoriniais ir eksperimentais su gyvūnais bei pakankamomis mokslinės literatūros žiniomis. Juos turi atlikti kvalifikuotas personalas, prižiūrimas patyrusio gydytojo. Visais atvejais gydytojas atsako už pacientą, bet ne patį pacientą, nepaisant jo duoto informuoto sutikimo.
Atliekant bet kokį tyrimą, kuriame dalyvauja žmonės, kiekvienas potencialus dalyvis turėtų būti tinkamai informuotas apie tyrimo tikslus, metodus, numatomą naudą ir susijusią riziką bei žalą. Žmonės turėtų būti informuoti, kad jie turi teisę susilaikyti nuo dalyvavimo tyrime ir gali atšaukti savo sutikimą bei atsisakyti tęsti tyrimą bet kuriuo metu po tyrimo pradžios. Tada gydytojas turi gauti savanoriškai duotą rašytinį informuotą tiriamojo sutikimą.
Kitas svarbus dokumentas, apibrėžiantis klinikinių tyrimų atlikimo etikos standartus, buvo „Tarptautinis biomedicininių tyrimų, kuriuose dalyvauja žmonės, etikos vadovas“ priimtas Tarptautinių medicinos mokslo organizacijų tarybos (CIOMS) (Ženeva, 1993 m.), kuriame pateikiamos gairės tyrėjams, finansuotojams, sveikatos apsaugos pareigūnams ir etikos komitetams, kaip įgyvendinti etikos standartus atliekant medicininius tyrimus, taip pat visiems svarbius etikos principus. asmenys, įskaitant klinikiniuose tyrimuose dalyvaujančius pacientus.
Helsinkio deklaracija ir Tarptautinės biomedicininių tyrimų, kuriuose dalyvauja žmonės, etikos gairės parodo, kaip pagrindiniai etikos principai gali būti veiksmingai taikomi medicinos tyrimų praktikoje visame pasaulyje, atsižvelgiant į skirtingas kultūrų, religijų, tradicijų, socialines ir ekonomines aplinkybes. sąlygas, įstatymus, administracines sistemas ir kitas situacijas, kurios gali susiklostyti ribotų išteklių šalyse.
1996 m. lapkričio 19 d. Europos Tarybos Parlamentinė Asamblėja priėmė „Žmogaus teisių ir žmogaus orumo apsaugos taikant biologiją ir mediciną konvencija“. Konvencijoje įtvirtintos normos turi ne tik moralinio kreipimosi galią – kiekviena prie jos prisijungusi valstybė įsipareigoja „pagrindines jos nuostatas įgyvendinti nacionalinėje teisėje“. Pagal šios Konvencijos nuostatas asmens interesai ir gerovė yra svarbesni už visuomenės ir mokslo interesus. Bet kokia medicininė intervencija, įskaitant intervenciją tyrimų tikslais, turi būti atliekama laikantis profesinių reikalavimų ir standartų. Tiriamasis turi iš anksto gauti atitinkamą informaciją apie intervencijos tikslą ir pobūdį, taip pat
jos pasekmės ir rizika; jo sutikimas turi būti savanoriškas. Medicininė intervencija asmeniui, negalinčiam duoti sutikimo, gali būti atliekama tik jo tiesioginiais interesais. 2005 m. sausio 25 d. buvo priimtas Papildomas Konvencijos protokolas dėl biomedicininių tyrimų.
Siekdama užtikrinti, kad būtų gerbiamos tiriamųjų teisės, tarptautinė bendruomenė šiuo metu sukūrė veiksmingą visuomenės ir vyriausybės kontrolės sistemą, užtikrinančią mokslinių tyrimų subjektų teises ir interesus bei klinikinių tyrimų etiką. Viena pagrindinių viešosios kontrolės sistemos grandžių yra nepriklausomų asmenų veikla etikos komitetai(EB).
Etikos komitetai šiandien yra struktūros, kurių požiūrio lauke susikerta moksliniai interesai, medicininiai faktai ir moralės bei teisės normos. Etikos komitetai atlieka tikrinimo, konsultavimo, rekomendacijų, skatinimo, vertinimo ir orientavimo funkcijas KI moralės ir teisės klausimais. Etikos komitetai atlieka labai svarbų vaidmenį nustatant, ar tyrimai yra saugūs, atliekami sąžiningai ir ar gerbiamos juose dalyvaujančių pacientų teisės; kitaip tariant, šie komitetai užtikrina visuomenei, kad kiekvienas atliktas klinikinis tyrimas atitinka etikos standartus.
EK turi būti nepriklausomos nuo tyrėjų ir neturi gauti materialinės naudos iš atliekamų tyrimų. Prieš pradėdamas darbą tyrėjas turi gauti komiteto patarimą, palankią apžvalgą arba leidimą. Komitetas vykdo tolesnę kontrolę, gali keisti protokolą ir stebėti tyrimo eigą bei rezultatus. Etikos komitetai turėtų turėti teisę uždrausti tyrimus, nutraukti tyrimus arba tiesiog atsisakyti patvirtinimo arba jį nutraukti.
Pagrindiniai etikos komitetų veiklos principai Atliekant etinį patikrinimą, KI yra nepriklausomumas, kompetencija, atvirumas, pliuralizmas, taip pat objektyvumas, konfidencialumas ir kolegialumas.
EK turi būti nepriklausomos nuo institucijų, priimančių sprendimus dėl klinikinių tyrimų vykdymo, įskaitant vyriausybines institucijas. Būtina komisijos kompetencijos sąlyga – aukšta jos protokolinės grupės kvalifikacija ir tikslus darbas (arba
sekretoriatas). Etikos komiteto veiklos atvirumą užtikrina jo darbo principų skaidrumas, nuostatai ir kt. Standartinės veiklos procedūros turėtų būti atviros visiems, kurie nori jas peržiūrėti. Etikos komiteto pliuralizmą garantuoja jos narių profesijų, amžiaus, lyčių ir religijų įvairovė. Peržiūros procese turi būti atsižvelgiama į visų tyrimo dalyvių, ypač ne tik pacientų, bet ir gydytojų, teises. Konfidencialumo reikalaujama dėl bylos medžiagos ir jame dalyvaujančių asmenų.
Nepriklausomas etikos komitetas dažniausiai kuriamas prie nacionalinių ar vietinių sveikatos departamentų, medicinos įstaigų ar kitų nacionalinių, regioninių, vietinių atstovaujamųjų organų pagrindu – kaip visuomeninė asociacija, nesudarant juridinio asmens.
Pagrindiniai etikos komiteto darbo tikslai yra tiriamųjų ir tyrėjų teisių ir interesų apsauga; nešališkas etinis klinikinių ir ikiklinikinių tyrimų (bandymų) vertinimas; kokybiškų klinikinių ir ikiklinikinių tyrimų (testų) atlikimo pagal tarptautinius standartus užtikrinimas; visuomenės pasitikėjimo, kad bus garantuoti ir laikomasi visų etikos principų.
Šiems tikslams pasiekti etikos komitetas turi spręsti šiuos uždavinius: savarankiškai ir objektyviai įvertinti žmogaus teisių saugumą ir vientisumą tiriamųjų atžvilgiu tiek planavimo, tiek tyrimo (testo) atlikimo stadijoje; įvertinti tyrimo atitiktį humanistiniams ir etiniams standartams, kiekvieno tyrimo (testo) pagrįstumą, tyrėjų atitikimą, technines priemones, tyrimo protokolą (programą), studijų dalykų atranką, atsitiktinės atrankos kokybę atlikimo taisyklėms. aukštos kokybės klinikiniai tyrimai; stebėti, kaip laikomasi klinikinių tyrimų kokybės standartų, kad būtų užtikrintas duomenų patikimumas ir išsamumas.
Rizikos ir naudos vertinimas yra svarbiausias etinis sprendimas, kurį EK priima peržiūrėdama mokslinių tyrimų projektus. Norint nustatyti, ar rizika yra pagrįsta, palyginti su nauda, reikia atsižvelgti į daugybę veiksnių ir kiekvieną atvejį reikėtų apsvarstyti atskirai,
atsižvelgiant į tyrime dalyvaujančių tiriamųjų (vaikų, nėščių moterų, nepagydomų ligonių) ypatumus.
Siekdama įvertinti riziką ir numatomą naudą, EK turi užtikrinti, kad:
Reikiamų duomenų negalima gauti neįtraukiant į tyrimą žmonių;
Tyrimas buvo racionaliai suplanuotas siekiant sumažinti tiriamųjų diskomfortą ir invazines procedūras;
Tyrimai padeda gauti svarbių rezultatų, skirtų diagnostikai ir gydymui tobulinti arba prisidėti prie duomenų apie ligas apibendrinimo ir sisteminimo;
Tyrimas pagrįstas laboratorinių duomenų ir eksperimentų su gyvūnais rezultatais, giliomis problemos istorijos žiniomis, o laukiami rezultatai tik patvirtins jos pagrįstumą;
Tikėtina tyrimo nauda viršija galimą riziką, o galima rizika yra minimali, t.y. ne daugiau kaip atliekant įprastines šios patologijos gydymo ir diagnostikos procedūras;
Tyrėjas turi pakankamai informacijos apie bet kokių galimų neigiamų tyrimo pasekmių nuspėjamumą;
Tiriamiesiems ir jų teisėtiems atstovams suteikiama visa informacija, reikalinga norint gauti jų informuotą ir savanorišką sutikimą.
Klinikiniai tyrimai turi būti atliekami pagal tarptautinių ir nacionalinių teisės aktų nuostatas, kurios garantuoja subjekto teisių apsauga.
Žmogaus teisių apsaugos konvencijos nuostatos gina asmens orumą ir asmens neliečiamybę bei kiekvienam be išimties garantuoja pagarbą asmens neliečiamumui ir kitoms teisėms bei pagrindinėms laisvėms, susijusioms su pažangos taikymu. biologija ir medicina, įskaitant transplantacijos, genetikos, psichiatrijos ir kt.
Jokie tyrimai su žmonėmis negali būti atliekami tuo pačiu metu nesilaikant visų šių sąlygų:
Nėra alternatyvių tyrimo metodų, kurių veiksmingumas būtų palyginamas;
Rizika, su kuria gali susidurti tiriamasis, neviršija galimos naudos iš šio tyrimo atlikimo;
Siūlomo tyrimo planą patvirtino kompetentinga institucija, atlikusi nepriklausomą mokslinio tyrimo privalumų, įskaitant jo tikslo svarbą, peržiūrą ir daugiašalį svarstymą dėl jo etinio priimtinumo;
Asmuo, veikiantis kaip subjektas, yra informuotas apie jo teises ir įstatymų numatytas garantijas;
Buvo gautas raštiškas sutikimas atlikti eksperimentą, kurį galima bet kada laisvai atšaukti.
„Rusijos Federacijos piliečių sveikatos apsaugos teisės aktų pagrindai“ ir Federalinis vaistų įstatymas „Dėl vaistų“ įtvirtina nuostatą, kad bet kokie biomedicininiai tyrimai, kuriuose dalyvauja žmogus kaip objektas, turi būti atliekami tik gavus raštišką Rusijos Federacijos piliečių sutikimą. pilietis. Žmogus negali būti verčiamas dalyvauti biomedicininiuose tyrimuose.
Gavęs sutikimą Norint atlikti biomedicininius tyrimus, piliečiui turi būti suteikta ši informacija:
1) apie vaistą ir jo klinikinių tyrimų pobūdį;
2) apie numatomą veiksmingumą, vaisto saugumą, rizikos laipsnį pacientui;
3) apie paciento veiksmus, atsiradus nenumatytam vaisto poveikiui jo sveikatai;
4) apie paciento sveikatos draudimo sąlygas.
Pacientas turi teisę atsisakyti dalyvauti klinikiniuose tyrimuose bet kuriame etape.
Informacija apie tyrimą pacientui turi būti perduota prieinama ir suprantama forma. Prieš gaudamas informuotą sutikimą, tyrėjas arba tyrėjas turi suteikti tiriamajam arba jo atstovui pakankamai laiko nuspręsti, ar dalyvauti tyrime, ir suteikti galimybę gauti išsamios informacijos apie tyrimą.
Informuotas sutikimas (informuotas paciento sutikimas) užtikrina, kad būsimi tiriamieji suprastų tyrimo pobūdį ir galėtų priimti informuotą bei savanorišką sprendimą
apie jūsų dalyvavimą ar nedalyvavimą. Ši garantija apsaugo visas puses: ir subjektą, kurio autonomija gerbiama, ir tyrėją, kuris kitaip pažeidžia įstatymus. Informuotas sutikimas yra vienas iš pagrindinių etikos reikalavimų atliekant tyrimus, kuriuose dalyvauja žmonės. Tai atspindi pagrindinį pagarbos asmenims principą. Informuoto sutikimo elementai apima visišką atskleidimą, tinkamą supratimą ir savanorišką pasirinkimą. Įvairios gyventojų grupės gali būti įtrauktos į medicininius tyrimus, tačiau draudžiama atlikti klinikinius vaistų tyrimus:
1) nepilnamečiai be tėvų;
2) nėščiosioms, išskyrus tuos atvejus, kai atliekami nėščioms moterims skirtų vaistinių preparatų klinikiniai tyrimai ir kai visiškai pašalinama rizika pakenkti nėščiajai ir vaisiui;
3) asmenys, atliekantys bausmę laisvės atėmimo vietose, taip pat esantys kardomojo kalinimo įstaigose be jų raštiško sutikimo.
Klinikinius vaistinių preparatų tyrimus su nepilnamečiais leidžiama atlikti tik tada, kai tiriamas vaistas skirtas išskirtinai vaikų ligoms gydyti arba kai klinikinio tyrimo tikslas – gauti duomenis apie geriausią vaisto dozę nepilnamečiams gydyti. Pastaruoju atveju prieš klinikinius tyrimus su vaikais turėtų būti atlikti panašūs tyrimai su suaugusiaisiais. Art. Rusijos Federacijos teisės aktų „Dėl piliečių sveikatos apsaugos“ pagrindų 43 straipsnyje pažymėta: „Gali būti naudojami diagnostikos, gydymo metodai ir vaistai, kurie nepatvirtinti vartoti, bet svarstomi nustatyta tvarka. jaunesniems nei 15 metų asmenims gydyti tik esant tiesioginei grėsmei jų gyvybei ir gavus raštišką jų teisėtų atstovų sutikimą. Informacija apie tyrimą vaikams turėtų būti perduota tokia kalba, kurią jie suprastų pagal amžių. Pasirašytą informuotą sutikimą gali gauti vaikai, sulaukę atitinkamo amžiaus (nuo 14 metų, kaip nustato įstatymai ir etikos komitetai).
Klinikinius vaistų, skirtų psichikos ligoms gydyti, tyrimus leidžiama atlikti su psichikos ligomis sergantiems asmenims ir asmenims, pripažintiems nekompetentingais.
nustatytas 1992 m. liepos 2 d. Rusijos Federacijos įstatymu Nr. 3185-1 „Dėl psichiatrinės pagalbos ir piliečių teisių garantijų ją teikiant“. Klinikiniai vaistinių preparatų tyrimai šiuo atveju atliekami gavus raštišką šių asmenų įstatyminių atstovų sutikimą.
3 skyrius. KLINIKINIAI VAISTŲ TYRIMAI
Prieš naujų vaistų atsiradimą vyksta ilgas tyrimų ciklas, kurių užduotis – įrodyti naujo vaisto veiksmingumą ir saugumą. Ikiklinikinių tyrimų su laboratoriniais gyvūnais principai buvo nusistovėję, tačiau praėjusio amžiaus ketvirtajame dešimtmetyje paaiškėjo, kad bandymų su gyvūnais rezultatai negali būti tiesiogiai perduodami žmonėms.
Pirmieji klinikiniai tyrimai su žmonėmis buvo atlikti XX amžiaus ketvirtojo dešimtmečio pradžioje (1931 m. – pirmasis atsitiktinių imčių aklas sanocrysin** tyrimas, 1933 m. 3 – pirmasis placebu kontroliuojamas tyrimas, kuriame dalyvavo pacientai, sergantys krūtinės angina). Šiuo metu visame pasaulyje yra atlikta keli šimtai tūkstančių klinikinių tyrimų (30 000-40 000 per metus). Prieš kiekvieno naujo vaisto pasirodymą atliekama vidutiniškai 80 skirtingų tyrimų, kuriuose dalyvavo daugiau nei 5000 pacientų. Tai žymiai pailgina naujų vaistų kūrimo laikotarpį (vidutiniškai 14,9 metų) ir reikalauja didelių išlaidų: vien klinikiniams tyrimams gamybos įmonės išleidžia vidutiniškai 900 mln. naujo vaisto veiksmingumas.vaistas.
Pagal tarptautines geros klinikinės praktikos gaires (tarptautinis klinikinių tyrimų standartas: ICH/GCP), pagal klinikinis tyrimas suprasti „tiriamojo vaisto saugumo ir (arba) veiksmingumo žmonėms tyrimą, kurio tikslas yra nustatyti arba patvirtinti klinikines, pageidaujamas tiriamojo vaisto farmakodinamines savybes ir (arba), siekiant nustatyti jo šalutinį poveikį ir (arba) ištirti jo absorbciją, pasiskirstymas, biotransformacija ir išskyrimas“.
Klinikinio tyrimo tikslas- gauti patikimų duomenų apie vaisto veiksmingumą ir saugumą neatskleidžiant
šiuo atveju pacientai (tyrimo subjektai) susiduria su nepagrįsta rizika. Konkrečiau, tyrimu gali būti siekiama ištirti vaisto farmakologinį poveikį žmogui, nustatyti terapinį (terapinį) efektyvumą arba patvirtinti veiksmingumą, palyginti su kitais vaistais, taip pat nustatyti terapinį panaudojimą – nišą, kurią šis vaistas gali užimti šiuolaikiniame pasaulyje. farmakoterapija. Be to, tyrimai gali būti vaisto paruošimo registracijai etapas, padėti reklamuoti jau registruotą vaistą rinkoje arba būti mokslo problemų sprendimo įrankiu.
3.1. KLINIKINIŲ TYRIMŲ STANDARTAI
Prieš sukuriant vienodus klinikinių tyrimų standartus, pacientai, vartojantys naujus vaistus, dažnai susidurdavo su rimta rizika, susijusia su nepakankamai veiksmingų ir pavojingų vaistų vartojimu. Pavyzdžiui, XX amžiaus pradžioje. daugelyje šalių heroinas buvo naudojamas kosuliui gydyti; 1937 m. JAV mirė kelios dešimtys vaikų, išgėrę paracetamolio sirupo, kuriame buvo toksiško etilenglikolio*; o septintajame dešimtmetyje Vokietijoje ir JK maždaug 10 000 vaikų gimė su sunkiais galūnių anomalijomis moterims, kurios nėštumo metu vartojo talidomidą*. Neteisingas tyrimų planavimas, rezultatų analizės klaidos ir atviri falsifikacijos sukėlė daugybę kitų humanitarinių nelaimių, dėl kurių kilo klausimas dėl tyrimuose dalyvaujančių pacientų ir galimų narkotikų vartotojų interesų apsaugos įstatymais.
Šiandien potenciali naujų vaistų skyrimo rizika yra žymiai mažesnė, nes vyriausybinės agentūros, duodančios sutikimą juos vartoti, turi galimybę įvertinti naujų vaistų vartojimo rezultatus tūkstančiams pacientų klinikinių tyrimų, atliekamų pagal vieną standartą, metu.
Šiuo metu visi klinikiniai tyrimai atliekami pagal vieną tarptautinį standartą, vadinamą GCP. , kurį sukūrė Vaistų kontrolės administracija
devintajame ir dešimtajame dešimtmetyje JAV vyriausybės, PSO ir Europos Sąjungos tiekimo ir maisto produktų. GCP standartas reglamentuoja klinikinių tyrimų planavimą ir vykdymą, taip pat numato kelių etapų pacientų saugos ir gautų duomenų tikslumo stebėseną.
GCP standarte atsižvelgiama į etikos reikalavimus moksliniams tyrimams, kuriuose dalyvauja žmonės, suformuluotus Pasaulio medikų asociacijos Helsinkio deklaracija„Rekomendacijos gydytojams, atliekantiems biomedicininius tyrimus, kuriuose dalyvauja žmonės“. Visų pirma, dalyvavimas klinikiniuose tyrimuose gali būti tik savanoriškas, pacientai neturėtų gauti piniginės kompensacijos tyrimo metu. Pacientas, pasirašydamas sutikimą tapti tyrimo dalyviu, gauna tikslią ir išsamią informaciją apie galimą pavojų jo sveikatai. Be to, pacientas gali bet kada, nenurodydamas priežasties, nustoti dalyvauti tyrime.
Kuriant GCP standartus ir visą šiuolaikinę vaistų klinikinių tyrimų koncepciją, didelę reikšmę turėjo klinikinė farmakologija, tirianti vaistų farmakokinetiką ir farmakodinamiką tiesiogiai sergančiam žmogui.
Tarptautinio standarto ICH GCP nuostatos atsispindi Federalinis įstatymas „Dėl vaistų apyvartos“(2010 m. balandžio 12 d. Nr. 61-FZ) ir Valstybinis standartas „Gera klinikinė praktika“(GOST R 52379-2005), pagal kurią mūsų šalyje atliekami klinikiniai vaistų tyrimai. Taigi yra teisinis pagrindas klinikinių tyrimų rezultatams tarp skirtingų šalių pripažinti, taip pat atlikti didelius tarptautinius klinikinius tyrimus.
3.2. KLINIKINIŲ TYRIMŲ PLANAVIMAS IR VYKDYMAS
Klinikinio tyrimo planavimas apima kelis etapus.
Tyrimo klausimo apibrėžimas. Pavyzdžiui, ar vaistas X reikšmingai sumažina kraujospūdį pacientams, sergantiems hipertenzija, ar vaistas X iš tikrųjų mažina kraujospūdį veiksmingiau nei vaistas Y? Tyrimą paprastai sudaro vienas pagrindinis tyrimas ir kelios papildomos tyrimo grupės.
klausimai, pavyzdžiui: ar vaistas Z gali sumažinti pacientų, sergančių hipertenzija, mirtingumą (pagrindinis klausimas), kaip vaistas Z veikia hospitalizavimo dažnumą, kokia vidutinio sunkumo hipertenzija sergančių pacientų dalis, kuriems vaistas Z gali patikimai kontroliuoti kraujospūdžio lygį (papildomi klausimai). Tyrimo klausimas atspindi prielaidą, nuo kurios tyrėjai pradeda (tyrimo hipotezė); mūsų pavyzdyje hipotezė yra ta, kad vaistas Z, turintis galimybę sumažinti kraujospūdį, gali sumažinti su hipertenzija susijusių komplikacijų ir ligų riziką, todėl gali sumažinti mirčių skaičių.
Studijų dizaino pasirinkimas. Tyrimas gali apimti keletą lyginamųjų grupių (vaistas A ir placebas arba vaistas A ir vaistas B). Lyginamosios grupės neturintys tyrimai patikimos informacijos apie vaistų poveikį nesuteikia, o šiuo metu tokie tyrimai praktiškai nevykdomi.
Imties dydžio nustatymas. Protokolo autoriai turi tiksliai numatyti, kiek pacientų reikės pirminei hipotezei įrodyti (imties dydis skaičiuojamas matematiškai, remiantis statistikos dėsniais). Tyrimas gali apimti nuo kelių dešimčių (tuo atveju, kai vaisto poveikis yra labai ryškus) iki 30 000-50 000 pacientų (jei vaisto poveikis yra silpnesnis).
Tyrimo trukmės nustatymas. Tyrimo trukmė priklauso nuo poveikio pradžios laiko. Pavyzdžiui, bronchus plečiantys vaistai ligonių, sergančių bronchine astma, būklę pagerina per kelias minutes po jų pavartojimo, tačiau registruoti teigiamą inhaliuojamųjų gliukokortikoidų poveikį šiems pacientams galima tik po kelių savaičių. Be to, kai kuriuose tyrimuose reikia stebėti gana retus reiškinius: jei tikimasi, kad tiriamasis vaistas sumažins ligos paūmėjimų skaičių, tai norint patvirtinti šį poveikį, būtinas ilgalaikis stebėjimas. Šiuolaikiniuose tyrimuose stebėjimo periodai svyruoja nuo kelių valandų iki 5-7 metų.
Pacientų populiacijos atranka. Norėdami į tyrimą įtraukti pacientus, turinčius tam tikrų savybių, kūrėjai sukuria aiškius kriterijus. Jie apima amžių, lytį, ligos trukmę ir sunkumą, ankstesnės ligos pobūdį
gydymas, gretutinės ligos, kurios gali turėti įtakos vaisto poveikio vertinimui. Įtraukimo kriterijai turėtų užtikrinti pacientų homogeniškumą. Pavyzdžiui, jei į hipertenzijos tyrimą vienu metu būtų įtraukti pacientai, sergantys lengva (ribine) hipertenzija, ir pacientai, kurių kraujospūdis labai aukštas, tiriamasis vaistas šiuos pacientus paveiktų skirtingai, todėl būtų sunku gauti patikimų rezultatų. Be to, į tyrimus dažniausiai neįtraukiamos nėščios moterys ir žmonės, sergantys sunkiomis ligomis, kurios neigiamai veikia bendrą paciento būklę ir prognozes.
Gydymo efektyvumo vertinimo metodai. Kūrėjai turi pasirinkti vaisto veiksmingumo rodiklius, mūsų pavyzdyje būtina išsiaiškinti, kaip tiksliai bus įvertintas hipotenzinis poveikis – vienu kraujospūdžio matavimu; apskaičiuojant vidutinį paros kraujospūdį; gydymo efektyvumas bus vertinamas pagal poveikį paciento gyvenimo kokybei arba pagal vaisto gebėjimą užkirsti kelią hipertenzijos komplikacijų apraiškoms.
Saugos vertinimo metodai. Turėtų būti taikomos priemonės gydymo saugumui įvertinti ir tiriamųjų vaistų NRV registravimo metodai.
Planavimo etapas baigiamas surašant protokolą – pagrindinį dokumentą, numatantį tyrimo atlikimą ir visas tyrimo procedūras. Taigi, tyrimo protokolas"apibūdina tyrimo tikslus, metodiką, statistinius aspektus ir organizavimą." Protokolas pateikiamas peržiūrėti vyriausybinėms reguliavimo institucijoms ir nepriklausomam etikos komitetui, be kurio sutikimo tyrimas negali prasidėti. Tyrimo vidinė (stebėsena) ir išorinė (auditas) kontrolė visų pirma vertina tyrėjų veiksmų atitiktį protokole aprašytai tvarkai.
Pacientų įtraukimas į tyrimą- grynai savanoriškai. Privaloma įtraukimo sąlyga yra tai, kad pacientas būtų supažindintas su galima rizika ir nauda, kurią jis gali gauti dalyvaudamas tyrime, taip pat pasirašydamas informuoto sutikimo. ICH GCP taisyklės neleidžia naudoti finansinių paskatų pritraukti pacientus dalyvauti tyrime (išimtis daroma sveikiems savanoriams, įdarbintiems tirti vaistų farmakokinetiką ar bioekvivalentiškumą). Pacientas turi atitikti įtraukimo / pašalinimo kriterijus. Paprastai
Nėščioms moterims, maitinančioms motinoms, pacientams, kuriems gali pakisti tiriamojo vaisto farmakokinetika, taip pat pacientams, sergantiems alkoholizmu ar priklausomybe nuo narkotikų, tyrimuose dalyvauti neleidžiama. Nepriimtina į tyrimą įtraukti neveiksnius pacientus be globėjų, karinio personalo, kalinių, asmenų, alergiškų tiriamajam vaistui, arba pacientų, kurie tuo pačiu metu dalyvauja kitame tyrime, sutikimo. Pacientas turi teisę bet kuriuo metu, nenurodydamas priežasčių, nustoti dalyvauti tyrime.
Studiju dizainas. Tyrimų, kurių metu visi pacientai būtų gydomi vienodai, šiuo metu praktiškai nėra, nes gautų rezultatų įrodymų nėra daug. Dažniausias lyginamasis tyrimas yra paralelinė grupė (intervencinė ir kontrolinė grupė). Kaip kontrolė gali būti naudojamas placebas (placebu kontroliuojamas tyrimas) arba kitas aktyvus vaistas.
Reikalingi tyrimai su lyginamuoju dizainu atsitiktinių imčių- atsitiktinis dalyvių paskirstymas į eksperimentines ir kontrolines grupes, kas leidžia sumažinti sistemines klaidas ir šališkumą. Tyrėjas iš esmės gali gauti informaciją apie tai, kokį vaistą pacientas vartoja (to gali prireikti, jei pasireiškia rimtos nepageidaujamos reakcijos), tačiau tokiu atveju pacientas turi būti pašalintas iš tyrimo.
Asmeninė registracijos kortelė. Individuali registracijos kortelė apibrėžiama kaip „spausdintas, optinis ar elektroninis dokumentas, sukurtas įrašyti visą protokolo reikalaujamą informaciją apie kiekvieną studijų dalyką“. Remiantis individualia registracijos kortele, sukuriama tyrimų duomenų bazė statistiniam rezultatų apdorojimui.
3.3. KLINIKINIŲ VAISTŲ BANDYMŲ ETAPAI
Tiek gamintojas, tiek visuomenė yra suinteresuoti gauti kuo tikslesnę ir išsamesnę informaciją apie naujo vaisto klinikinę farmakologiją, terapinį efektyvumą ir saugumą išankstinės registracijos tyrimų metu. Paruošimas
registracijos dokumentacija neįmanoma be atsakymų į šiuos klausimus. Dėl šios priežasties prieš registruojant naują vaistą atliekamos kelios dešimtys skirtingų tyrimų, kasmet didėja tiek tyrimų, tiek jų dalyvių skaičius, o bendras naujo vaisto tyrimų ciklas paprastai viršija 10 metų. Taigi naujų vaistų kūrimas įmanomas tik didelėse farmacijos įmonėse, o bendra tyrimo projekto kaina vidutiniškai viršija 900 mln.
Pirmieji ikiklinikiniai tyrimai prasideda netrukus po naujos, potencialiai veiksmingos molekulės sintezės. Jų esmė – patikrinti hipotezę apie numatomą naujo junginio farmakologinį poveikį. Tuo pačiu metu tiriamas junginio toksiškumas, jo onkogeninis ir teratogeninis poveikis. Visi šie tyrimai atliekami su laboratoriniais gyvūnais, o bendra jų trukmė – 5-6 metai. Dėl šio darbo iš 5-10 tūkstančių naujų junginių atrenkama maždaug 250.
Patys klinikiniai tyrimai paprastai skirstomi į keturis laikotarpius arba fazes.
I fazės klinikiniai tyrimai, paprastai atliekama 28–30 sveikų savanorių. Šio etapo tikslas – gauti informaciją apie naujo vaisto toleravimą, farmakokinetiką ir farmakodinamiką, patikslinti dozavimo režimą ir gauti duomenis apie vaisto saugumą. Šiame etape nebūtina tirti terapinio vaisto poveikio, nes sveikiems savanoriams kai kurios kliniškai svarbios naujojo vaisto savybės paprastai nepastebimos.
I fazės tyrimai pradedami nuo vienos dozės saugumo ir farmakokinetikos tyrimo, kurios parinkimas grindžiamas duomenimis, gautais iš biologinių modelių. Ateityje tiriama vaisto farmakokinetika vartojant kartotinį, naujo vaisto išsiskyrimas ir metabolizmas (kinetinių procesų tvarka), jo pasiskirstymas skysčiuose ir kūno audiniuose, farmakodinamika. Paprastai visi šie tyrimai atliekami naudojant skirtingas dozes, dozavimo formas ir vartojimo būdus. I fazės tyrimų metu taip pat galima įvertinti kitų vaistų, organizmo funkcinės būklės, suvartojamo maisto ir kt. poveikį naujo vaisto farmakokinetikai ir farmakodinamikai.
Svarbus I fazės klinikinių tyrimų tikslas yra nustatyti galimą toksiškumą ir NRV, tačiau šie tyrimai yra trumpalaikiai ir atliekami ribotam dalyvių skaičiui, todėl per šį etapą galima nustatyti tik labiausiai
dažni ir sunkūs nepageidaujami reiškiniai, susiję su naujų vaistų vartojimu.
Kai kuriais atvejais (onkologiniai vaistai, vaistai ŽIV infekcijai gydyti) pacientams gali būti atliekami I fazės tyrimai. Tai leidžia paspartinti naujo vaisto kūrimą ir nesukelti savanorių nepagrįstos rizikos, nors šį metodą galima laikyti labiau išimtimi.
I pakopos studijos leisti:
Įvertinti naujo vaisto toleravimą ir saugumą;
Kai kuriais atvejais susidarykite supratimą apie jo farmakokinetiką (sveikiems žmonėms, o tai natūraliai turi ribotą reikšmę);
Nustatykite pagrindines farmakokinetines konstantas (Cmax,
C1);
Palyginkite naujo vaisto farmakokinetiką, naudodami skirtingas dozavimo formas, būdus ir vartojimo būdus.
II fazės tyrimai- pirmieji tyrimai su pacientais. Šių tyrimų apimtis yra žymiai didesnė nei I fazėje: 100-200 pacientų (kartais iki 500). II fazėje aiškinamasi naujojo vaisto veiksmingumas ir saugumas bei pacientų gydymui skirtų dozių diapazonas. Šie tyrimai suteikia informacijos daugiausia apie naujojo vaisto farmakodinamiką. Lyginamasis planas ir kontrolinės grupės įtraukimas yra laikomi privalomomis II fazės tyrimų atlikimo sąlygomis (tai nebūdinga I fazės tyrimams).
III fazės tyrimai planuojami dideliam pacientų skaičiui (iki 10 000 ir daugiau žmonių), o jų įgyvendinimo sąlygos yra kuo artimesnės įprastoms tam tikrų ligų gydymo sąlygoms. Šio etapo tyrimai (dažniausiai keli lygiagretūs arba nuoseklūs tyrimai) yra dideli (viso masto), atsitiktinių imčių ir lyginamieji. Tyrimo objektas yra ne tik naujojo vaisto farmakodinamika, bet ir jo klinikinis efektyvumas 1 .
1 Pavyzdžiui, naujo antihipertenzinio vaisto I-II fazėse tyrimo tikslas – įrodyti jo gebėjimą mažinti kraujospūdį, o III fazės tyrimo tikslas – ištirti vaisto poveikį hipertenzijai. Pastaruoju atveju, kartu su kraujospūdžio sumažėjimu, atsiranda ir kiti poveikio vertinimo taškai, ypač mirtingumo nuo širdies ir kraujagyslių ligų sumažėjimas, hipertenzijos komplikacijų prevencija, pacientų gyvenimo kokybės gerinimas ir kt.
III fazės tyrimuose vaisto veiksmingumas ir saugumas lyginamas su placebu (placebu kontroliuojamas tyrimas) ir (arba) su kitu žymekliu (vaistu, kuris dažniausiai naudojamas tam tikroje klinikinėje situacijoje ir turintis gerai žinomų vaistinių savybių).
Tai, kad kūrėjas pateikia paraišką registruoti vaistus, nereiškia, kad tyrimas yra baigtas. III fazės tyrimai, atlikti prieš pateikiant paraišką, vadinami Ia fazės tyrimais, o po paraiškos padavimo – III fazės tyrimais. Pastarosios atliekamos siekiant gauti išsamesnės informacijos apie klinikinį ir farmakoekonominį vaistų efektyvumą. Tokie tyrimai gali išplėsti naujo vaisto skyrimo indikacijas. Papildomus tyrimus gali inicijuoti už registravimo procesą atsakingos vyriausybinės agentūros, jei ankstesnių tyrimų rezultatai neleidžia vienareikšmiškai teigti apie naujojo vaisto savybes ir saugumą.
III fazės tyrimų rezultatai tampa lemiami priimant sprendimą registruoti naują vaistą. Toks sprendimas gali būti priimtas, jei vaistas:
Veiksmingesni nei jau žinomi panašaus veikimo vaistai;
Turi poveikį, kuris nėra būdingas esamiems vaistams;
Turi palankesnę dozavimo formą;
Farmakoekonomine prasme naudingesnis arba leidžia naudoti paprastesnius gydymo metodus;
Jis turi pranašumų, kai naudojamas kartu su kitais vaistais;
Turi paprastesnį naudojimo būdą.
IV fazės tyrimai. Konkurencija su naujais vaistais verčia tęsti tyrimus net ir įregistravus naują vaistą (tyrimai po pateikimo į rinką), siekiant patvirtinti vaisto veiksmingumą ir vietą farmakoterapijoje. Be to, IV fazės tyrimai leidžia atsakyti į kai kuriuos klausimus, kylančius vartojant vaistus (optimali gydymo trukmė, naujo vaisto privalumai ir trūkumai, lyginant su kitais, įskaitant naujesnius vaistus, recepto ypatumai vyresnio amžiaus žmonėms , vaikai, ilgalaikis gydymo poveikis, naujos indikacijos ir kt.).
Kartais IV fazės tyrimai atliekami praėjus daugeliui metų po vaisto registracijos. Tokių daugiau nei 60 metų atidėjimų pavyzdys
Visų fazių klinikiniai tyrimai atliekami 2 valstybinės kontrolės institucijų oficialiai sertifikuotuose centruose (medicinos centruose, ligoninėse, klinikose), turinčiuose atitinkamą mokslinę ir diagnostinę įrangą bei galimybę suteikti kvalifikuotą medicininę pagalbą pacientams, sergantiems NRV.
Bioekvivalentiškumo tyrimai. Dauguma vaistų rinkoje yra atkurti (generiniai) vaistai. Į šiuos vaistus įtrauktų vaistų farmakologinis poveikis ir klinikinis veiksmingumas, kaip taisyklė, buvo gana gerai ištirtas. Tačiau generinių vaistų veiksmingumas gali labai skirtis.
Generinių vaistų registracija gali būti supaprastinta (atsižvelgiant į tyrimų laiką ir apimtį). Bioekvivalentiškumo tyrimai leidžia daryti griežtai pagrįstą išvadą apie šių produktų kokybę. Šiuose tyrimuose generinis vaistas lyginamas su pirminiu vaistu pagal biologinį prieinamumą (lyginama vaisto, patenkančio į sisteminę kraujotaką, dalis ir šio proceso greitis). Jei dviejų vaistų biologinis prieinamumas yra toks pat, jie yra biologiškai ekvivalentiški. Daroma prielaida, kad bioekvivalentiški vaistai turi tokį patį veiksmingumą ir saugumą 3 .
Bioekvivalentiškumas tiriamas su nedideliu sveikų savanorių skaičiumi (20–30), naudojant standartines farmakokinetikos tyrimo procedūras (farmakokinetinės kreivės sudarymas, AUC, Tmax, Cmax verčių tyrimai).
maks maks
1 Šie vaistai klinikinėje praktikoje buvo pasiūlyti maždaug prieš 100 metų, bet vienu metu nebuvo registruojami ir nebuvo tiriami klinikiniai tyrimai, todėl daugiau nei po 60 metų reikėjo atlikti išsamius tyrimus. Šiuolaikinė naujų vaistų registravimo sistema atsirado XX amžiaus šeštajame dešimtmetyje, todėl apie 30–40% šiandien vartojamų vaistų nėra įtikinamai ištirta. Jų vieta farmakoterapijoje gali būti diskusijų objektas. Literatūroje anglų kalba šiems vaistams vartojamas terminas „retieji vaistai“, nes retai pavyksta rasti finansavimo šaltinių tokių vaistų tyrimams.
2 Mūsų šalyje - Rusijos Federacijos sveikatos ir socialinės plėtros ministerija.
3 Tačiau negalima teigti, kad du farmaciniu požiūriu lygiaverčiai vaistai (to paties veiksmingumo ir saugumo) visada turi tą pačią farmakokinetiką ir panašų biologinį prieinamumą.
3.4. ETINIAI KLINIKINIAI ASPEKTAI
TYRIMAI
Svarbiausias medicinos etikos principas buvo suformuluotas beveik prieš 2500 metų. Hipokrato priesaikoje teigiama: „Aš įsipareigoju visa tai daryti pagal savo sugebėjimus ir žinias paciento labui ir susilaikyti nuo visko, kas gali pakenkti jam“. Medicininės deontologijos reikalavimai įgyja ypatingą reikšmę atliekant klinikinius vaistų tyrimus, nes jie atliekami su žmonėmis ir daro įtaką žmogaus teisėms į sveikatą ir gyvybę. Vadinasi, medicininės-teisinės ir medicininės-deontologinės problemos turi didelę reikšmę klinikinėje farmakologijoje.
Atliekant klinikinius vaistų tyrimus (tiek naujų, tiek jau ištirtų, bet vartojamų naujoms indikacijoms), pirmiausia reikia vadovautis paciento interesais. Kompetentingos institucijos (Rusijos Federacijoje - Rusijos sveikatos ir socialinės plėtros ministerija) sutinka atlikti klinikinius vaistų tyrimus, išsamiai išnagrinėjusios ikiklinikinio vaisto tyrimo metu gautų duomenų visumą. Tačiau, nepaisant vyriausybės patvirtinimo, tyrimas taip pat turi gauti etikos komiteto patvirtinimą.
Klinikinių tyrimų etinė peržiūra atliekama pagal Pasaulio medikų asociacijos Helsinkio deklaracijos „Rekomendacijos gydytojams, atliekantiems biomedicininius tyrimus, kuriuose dalyvauja žmonės“ principus (pirmą kartą priimta 18-ojoje Pasaulio medicinos asamblėjoje Helsinkyje 1964 m., o vėliau). keletą kartų taisytas ir peržiūrėtas).
Helsinkio deklaracijoje teigiama, kad biomedicininių tyrimų su žmonėmis tikslas turėtų būti diagnostinių, terapinių ir prevencinių procedūrų tobulinimas, taip pat ligų etiologijos ir patogenezės išaiškinimas. Pasaulio medicinos asamblėja parengė rekomendacijas gydytojams atliekant klinikinius tyrimus.
Į Helsinkio deklaracijos reikalavimus buvo atsižvelgta Rusijos Federacijos federaliniame įstatyme „Dėl vaistų apyvartos“. Visų pirma tai patvirtina įstatymas.
Pacientų dalyvavimas klinikiniuose vaistų tyrimuose gali būti tik savanoriškas.
Pacientas duoda raštišką sutikimą dalyvauti klinikiniuose vaistų tyrimuose.
Pacientas turi būti informuotas apie tyrimo pobūdį ir galimą pavojų jo sveikatai.
Pacientas turi teisę atsisakyti dalyvauti klinikiniuose vaistų tyrimuose bet kuriame etape.
Remiantis etikos reikalavimais, klinikiniai vaistų tyrimai, susiję su nepilnamečiais (išskyrus tuos atvejus, kai tiriamas vaistas skirtas išskirtinai vaikų ligoms gydyti) ir nėščiosioms, yra draudžiami. Draudžiama atlikti klinikinius narkotikų tyrimus nepilnamečiams be tėvų, nekompetentingiems asmenims, kaliniams, kariškiams ir kt. Visi klinikinių tyrimų dalyviai turi būti apdrausti.
Klinikinių tyrimų etinės peržiūros klausimus mūsų šalyje sprendžia Rusijos sveikatos ir socialinės plėtros ministerijos etikos komitetas, taip pat vietiniai etikos komitetai prie medicinos ir mokslo medicinos įstaigų. Etikos komitetas vadovaujasi pagrindiniais tarptautiniais klinikinių tyrimų atlikimo principais, taip pat galiojančiais Rusijos Federacijos įstatymais ir reglamentais.
3.5. NAUJŲ VAISTŲ REGISTRAVIMO TVARKA
Pagal federalinį įstatymą „Dėl vaistų apyvartos“ (2010 m. balandžio 12 d. Nr. 61-FZ) „Vaistai gali būti gaminami, parduodami ir naudojami Rusijos Federacijos teritorijoje, jei jie yra įregistruoti federalinėje įstaigoje. vaistų kokybės kontrolė“. Valstybinė registracija privaloma:
Nauji vaistai;
Nauji anksčiau registruotų vaistų deriniai;
Vaistai, registruoti anksčiau, bet pagaminti kitomis dozavimo formomis arba naujomis dozėmis;
Generiniai vaistai.
Valstybinę vaistų registraciją vykdo Rusijos sveikatos ir socialinės plėtros ministerija, ji taip pat tvirtina vaistų vartojimo instrukcijas, o registruotas vaistas įtraukiamas į valstybinį registrą.
Klinikinė farmakologija ir farmakoterapija: vadovėlis. - 3 leidimas, pataisytas. ir papildomas / red. V. G. Kukesa, A. K. Starodubceva. - 2012. - 840 p.: iliustr.
1. Medicinos reikmėms skirtų vaistų klinikiniai tyrimai, įskaitant tarptautinius daugiacentrius, daugiacentrius, poregistracinius, atliekami vienoje ar keliose medicinos organizacijose pagal geros klinikinės praktikos taisykles, kurias atitinkamai patvirtino įgaliota federalinė vykdomoji institucija. šiais tikslais:
1) nustatantis vaistų, skirtų sveikiems savanoriams, saugumą ir (ar) jų toleravimą sveikiems savanoriams, išskyrus tokius tyrimus su vaistiniais preparatais, pagamintais už Rusijos Federacijos ribų;
3) nustatant vaisto saugumą ir veiksmingumą sergant tam tikra liga, imunobiologinių vaistų profilaktinį veiksmingumą sveikiems savanoriams;
4) tiria galimybę išplėsti medicininio vartojimo indikacijas ir nustatyti anksčiau nežinomus registruotų vaistų šalutinius poveikius.
2. Genetinių medicinos reikmėms skirtų vaistų bioekvivalentiškumo ir (ar) terapinio lygiavertiškumo tyrimai atliekami įgaliotos federalinės vykdomosios institucijos nustatyta tvarka.
3. Organizuoti medicininio preparato klinikinius tyrimus turi teisę:
1) vaisto kūrėjas arba jo įgaliotas asmuo;
2) aukštojo mokslo švietimo organizacijos, papildomo profesinio rengimo organizacijos;
(žr. tekstą ankstesniame leidime)
3) mokslinių tyrimų organizacijos.
4. Medicinos paskirties vaistinio preparato klinikiniai tyrimai atliekami pagal įgaliotos federalinės vykdomosios institucijos išduotą leidimą atlikti klinikinį vaistinio preparato tyrimą. Įgaliota federalinė vykdomoji institucija šios institucijos nustatyta tvarka tvarko išduotų leidimų atlikti klinikinius vaistinio preparato tyrimus registrą, kuriame nurodoma jų paskirtis ar tikslai.
(žr. tekstą ankstesniame leidime)
(žr. tekstą ankstesniame leidime)
6. Organizuojant vaistinio preparato, skirto medicinos reikmėms, klinikinius tyrimus, kuriuos atlieka vaisto kūrėjas, gali dalyvauti bet kokios organizacinės ir teisinės formos juridiniai asmenys, jeigu šie tyrimai atitinka šio federalinio įstatymo reikalavimus.
7. Medicinos reikmėms skirtų vaistų klinikiniai tyrimai atliekami medicinos organizacijose, kurias Rusijos Federacijos Vyriausybės nustatyta tvarka akreditavo įgaliota federalinė vykdomoji institucija.
8. Medicinos organizacijų, turinčių teisę atlikti medicininių vaistų klinikinius tyrimus, sąrašą ir išduotų leidimų atlikti klinikinius vaistinių preparatų tyrimus registrą skelbia ir paskelbia įgaliota federalinė vykdomoji institucija savo nustatyta tvarka. savo oficialioje svetainėje internete.

2018 metais gimimo pasninkas prasidės lapkričio 28 d. Šiuo laikotarpiu stačiatikiai ruošiasi švęsti Kalėdas...

Daugumos moterų svajonė yra sukurti šeimą. Jie nori turėti mylintį vyrą ir krūvą vaikų. Bet tai ne visada yra santykiai...

Šiame straipsnyje pateikiama: galingiausia skyrybų malda – informacija, paimta iš viso pasaulio, elektroninio tinklo ir...

Informacinė svetainė apie ikonas, maldas, stačiatikių tradicijas. Malda už skandalus ir kivirčus šeimoje, su vyru, su vaikais...

Kokie būtų Naujieji metai be šampano, mandarinų, Olivier, aspicų ir visų mėgstamo „Silkės po kailiu“. Su paskutiniu...

Paruoškime sausainiams reikalingus ingredientus. Pirmas dalykas, kurį reikia padaryti, yra užvirti vandenį. Mums reikia...

Ar galima registruoti darbuotoją į finansų direktoriaus – vyriausiojo buhalterio pareigas? Vyriausioji buhalterė tvirtina...

Smulkaus verslo vadovas gali lengvai valdyti biudžetą savarankiškai. PATIKRINTA! Jei pavyks...

Naujų projektų kūrimas apima preliminarų ekonominį jų pagrįstumo pagrindimą, vėlesnius...
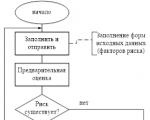
Ataskaitas rengia RM, dėl jų susitaria (patvirtina) Rizikos komitetas prie valdybos ir perduoda...

Didelės, labai didelės pievos pakraštyje, ant ilgo smaragdinio žolės stiebo gyveno mažytė boružėlė. Dievo mažutė...

Šiais laikais gana įprasta, kad žmonės atsigręžia į žvaigždes. Horoskopo pagalba žmogus gali sužinoti...

Verslo ar draugiško. Jei persekiojote nepažįstamą žmogų, tai reiškia jūsų pasitikėjimo moteriškąja lytimi lygį...

pagal Freudo svajonių knygą Jei svajojote, kaip žvejojote, tai reiškia, kad realiame gyvenime vargu ar galite išsijungti...

Daugumos moterų svajonė yra sukurti šeimą. Jie nori turėti mylintį vyrą ir krūvą vaikų. Bet ne visada...

Šiame straipsnyje yra: pati galingiausia malda už skyrybas – informacija paimta iš viso pasaulio, elektroninė...