O que é possível e o que não é possível durante o Jejum da Natividade?
Em 2018, o Jejum da Natividade começará em 28 de novembro. Durante este período, os crentes ortodoxos se preparam para celebrar o Natal...
O quadro clínico da doença foi descrito pela primeira vez por Andersen em 1956. A doença é herdada de forma autossômica recessiva. Na glicogenose tipo IV, há um defeito na enzima amilo-1,4 → 1,6-transglucosidase, que está envolvida na formação de pontos de ramificação na molécula de glicogênio:
Na glicogenose tipo IV, o glicogênio anormal, semelhante à amilopectina (um componente do amido nas células vegetais), é sintetizado nos órgãos afetados. A molécula de glicogênio anormal tem um número reduzido de pontos de ramificação e cadeias externas e internas mais longas em comparação com a norma.
A doença é rara e generalizada (os músculos cardíacos, esqueléticos e o fígado são os mais afetados). Clinicamente, a doença se manifesta por hepatoesplenomegalia, ascite, o desenvolvimento mental não é afetado. A fibrose portal progressiva do fígado leva à cirrose. A cirrose provavelmente se desenvolve como resultado do acúmulo de glicogênio semelhante à amila.
Morte na infância por insuficiência hepática. Um exame anatomopatológico revela um aumento no tamanho dos rins, fígado e baço. Os hepatócitos aumentam de tamanho e contêm polissacarídeo semelhante à amilopectina.
Doença de Lafora- glicogenose cerebral (epilepsia mioclônica). Nesta doença, é detectado um acúmulo de glicogênio anormal no cérebro, semelhante às propriedades do polímero na glicogenose tipo IV. A atividade da enzima ramificada não é alterada nesta doença.
Descrito pela primeira vez por B. McArdle em 1951. Tipo de herança autossômica recessiva. Caracterizado pela deficiência de fosforilase muscular nos músculos esqueléticos. A ausência de fosforilase muscular não está combinada com uma violação da fosforilase hepática (controlada por vários genes). A atividade da fosforilase de leucócitos, eritrócitos e plaquetas na doença de McArdle não é alterada.
Com esta doença, até 3-4% do glicogênio de estrutura normal se acumula nas fibras musculares. O excesso de glicogênio é depositado sob o sarcolema no citoplasma. Em repouso, as necessidades energéticas são supridas pela glicose dos miócitos. Durante o trabalho muscular, a necessidade de fornecimento de energia não é atendida devido a um defeito enzimático, que causa dores e cãibras neste tipo de glicogenose.
A doença de McArdle é heterogênea. Os sinais clínicos aparecem com mais frequência em adultos, na infância os sintomas da doença não são pronunciados. A doença ocorre em 3 fases:
1. Na infância e adolescência são observadas fraqueza muscular, fadiga e possível mioglobinúria.
2. Entre os 20 e os 40 anos, as dores musculares tornam-se mais intensas e surgem cãibras após a atividade física.
3. Após 40 anos, ocorre fraqueza progressiva devido à distrofia muscular.
Foi estabelecido que a atividade da fosforilase diminui acentuadamente com a deficiência de vitamina B6 (60% da piridoxina nos músculos esqueléticos está associada à fosforilase). Portanto, a deficiência de fosforilase afeta o conteúdo de piridoxina no organismo. O prognóstico para glicogenose tipo V é favorável.
Uma doença hereditária rara do grupo das canalopatias multissistêmicas. O modo de herança é autossômico dominante, com transição incompleta e variabilidade significativa entre membros da mesma família. Casos esporádicos são comuns. O gene defeituoso (KCNJ2) está localizado no braço longo do cromossomo 17 (locus 17q23.1-q24.2). O produto genético está envolvido na formação de canais de potássio, através dos quais o potássio entra nas células musculares. Quando ocorre uma mutação genética, a estrutura dos canais de potássio é perturbada, assim como a regulação da entrada de íons potássio na célula (a molécula reguladora PIP2 não consegue entrar em contato com o canal). A penetração prejudicada de íons potássio nas células musculares leva ao desenvolvimento de sinais característicos da síndrome (o papel do gene KCNJ2 na formação do sistema esquelético ainda está sendo estudado). Clinicamente, a síndrome é representada por uma tríade de sinais:
dismorfismo característico da face e do esqueleto;
paralisia periódica sensível ao potássio;
arritmias ventriculares.
Danos ao aparelho valvular do coração e hipoplasia renal também são possíveis.
As características displásicas são representadas por baixa estatura, orelhas de implantação baixa, hipertelorismo, defeitos do palato mole e duro, hipoplasia do maxilar inferior, clinodactilia e escoliose.
O rosto de um paciente com síndrome de Andersen-Tawil. Destacam-se os traços displásicos característicos: hipertelorismo, hipoplasia do maxilar inferior e orelhas de inserção baixa (fonte Katz J.S., Wolfe GI, Iannaccone S., Bryan W.W., Barohn RJ O teste de exercício na síndrome de Andersen // Arch. Neurol., 1999. – Vol.56. – P.352-356)
A paralisia periódica sensível ao potássio sem manifestações miotônicas características desta síndrome é clinicamente indistinguível de outras formas de paralisia periódica hipercalêmica. No entanto, existe a opinião de que, devido à extrema variabilidade nas alterações nas concentrações de potássio durante as crises paralíticas, os critérios tradicionais para as formas hipo, normo e hipercalêmica na síndrome de Andersen-Tawil são inaceitáveis. Freqüentemente, os ataques se desenvolvem num contexto de fraqueza geral prolongada.
Os sintomas cardíacos incluem prolongamento do intervalo QT de gravidade variável, bigeminismo ventricular, taquicardia ventricular paroxística (até biventricular), parada cardíaca súbita.
Existem relatos na literatura de síndrome de morte súbita em pacientes portadores desta doença.
Os pacientes frequentemente apresentam reações paradoxais à administração de vários medicamentos e refratariedade aos antiarrítmicos. Foi demonstrado um efeito positivo persistente da terapia com amiodarona e acetazolamida (Diacarb) (alívio dos sintomas cardíacos e musculares).
Pela primeira vez, a combinação de paralisia periódica e arritmia foi notada por Klein et al. em 1963 ( Klein R., Ganelin R., Marks J.F., Usher P., Richards C. Paralisia periódica com arritmia cardíaca // J. Pediatr., 1963. – Vol.62. – P.371-385) e Lisak et al. em 1970 ( Lisak RP, Lebeau J., Tucker SH, Rowland LP. Paralisia periódica hipercalêmica com arritmia cardíaca // Neurologia, 1970. – Vol.20. –Pág.386). A síndrome foi descrita pela primeira vez pela médica dinamarquesa Ellen Damgaard Andersen e coautores em 1971. ( Andersen E. D., Krasilnikoff P. A., Overvad H. Intermitente muscular fraqueza, extra-sístoles e múltiplo desenvolvimento anormalidades: a novo síndrome? // Acta pediátrica Escandinavo, Estocolmo, 1971. – Vol..60. – P.559–564 ); descreveu um caso da doença em uma criança de 8 anos com tríade característica de paralisia periódica, arritmia e anomalias de desenvolvimento. Posteriormente, tal tríade foi descrita apenas em um único trabalho em 1985. E apenas uma descrição detalhada do neurologista americano de origem libanesa, Rabi Tawil et al. ( Tawil R., Ptacek eu. J.., Pavlakis S. G., DeVivo D. C., Penn A. S., Özdemir C., Griggs R. C. Andersen‘ é síndrome: potássio— confidencial periódico paralisia, ventricular ectopia, e dismórfico características // Anuais de Neurologia, 1994. – Vol..35. – N.3. – P.326-330 ), atraiu a atenção de especialistas para essa forma nosológica, estimulando seu aprofundamento.
13. Priori SG, Napolitano C., Grillo M. Síndromes arritmogênicas ocultas: O substrato oculto da fibrilação ventricular idiopática? // Cardiovasc. Res. - 2001. - Vol. 50.
14. Priori S. G., Napolitano C., Memmi M. Caracterização clínica e molecular de pacientes com taquicardia ventricular polimórfica catecolaminérgica // Circulação. - 2002.
Vol. 106. - S. 69-74.
15. Priori SG, Napolitano C., Tiso N. et al. Mutações no gene do receptor de rianodina cardíaco (hRyR2) estão subjacentes à taquicardia ventricular polimórfica catecolaminérgica // Ibid.
2000. -Vol. 103. - S. 196-200.
16. Spurgeon D. Mortes cardíacas súbitas aumentam 10% em jovens americanos // Brit. Med. J. - 2001. - Vol. 322. - P. 573 (Resumo).
17. Sumitomo N., Harada K., Nagashima M. et al. Taquicardia ventricular polimórfica catecolaminérgica:
© SM KRUPIANKO, TT KAKUCHAYA, 2005 UDC 616.12-008.6
SÍNDROME DE ANDERSEN
SM Krupyanko, TT Kakuchaya
Centro Científico de Cirurgia Cardiovascular em homenagem. A.
RAMS, Moscou
A síndrome de Andersen é uma patologia hereditária rara caracterizada por paralisia muscular transitória, prolongamento do intervalo QT, muitas vezes combinado com o aparecimento de ondas U de alta amplitude, arritmias ventriculares e sinais de dismorfogênese - orelhas de implantação baixa, micrognatia (mandíbulas anormalmente pequenas, especialmente maxilar inferior), testa larga, clinodactilia (deformação persistente de um ou mais dedos), sindactilia (fusão ou presença de membranas entre os dedos das mãos ou dos pés), hipertelorismo (aumento da distância entre dois órgãos pares), baixa estatura, escoliose , etc. Em 1971, E. Andersen et al. relataram que um paciente de 8 anos tinha baixa estatura, hipertelorismo (olhos bem espaçados), hipoplasia da mandíbula, base larga do nariz, fenda palatina, crânio escafocefálico (um crânio longo e estreito com uma crista ao longo da sutura sagital ossificada), e clinodactilia do quinto dígito. Em 1994, R. Tawil et al. utilizou pela primeira vez o termo “síndrome de Andersen” para descrever um caso clínico com três aspectos característicos (tríade clínica): paralisia cíclica sensível ao potássio, distúrbios do ritmo ventricular e sinais de dismorfogênese, observados por Andersen em 1971. A literatura também contém frequentemente a definição “síndrome de Andersen-Tawil” (síndrome de Andersen-Tawil).
Características eletrocardiográficas e estratégias terapêuticas ótimas para prevenir morte súbita // Coração. - 2003.
Vol. 89, nº 1. - P. 66-70.
18. Swan H, Laitinen PJ, Kontula K. et al. O antagonismo dos canais de cálcio reduz arritmias ventriculares induzidas por exercício em pacientes com taquicardia ventricular polimórfica catecolaminérgica com mutações RYR2 // J. Cardiovasc. Eletrofisiol. - 2005. - Vol. 16, nº 2. - P. 162-166 (Resumo).
19. Swan H., Piippo K., Viitasalo M. et al. Distúrbio arrítmico mapeado no cromossomo 1q42-q43 causa taquicardia ventricular polimórfica maligna em corações estruturalmente normais // J. Amer. Col. Cardiol. - 1999. - Vol. 34, nº 7.
20. Tan H.L., Hofman N., Van Langen I.M. et al. Morte súbita e inexplicável. Herdabilidade e rendimento diagnóstico do exame cardiológico e genético em parentes sobreviventes // Circulação. - 2005. - Vol. 112. - S. 207-213.
N. Bakuleva (diretor - Acadêmico da Academia Russa de Ciências Médicas L. A. Bokeria)
drome, abreviado como ATS). Esta síndrome não deve ser confundida com a doença de Andersen, que pertence à glicogenose - doenças do armazenamento de glicogênio (devido à deficiência da enzima conversora de glicogênio, uma quantidade patológica de glicogênio se acumula no fígado, músculos e outros tecidos). A síndrome de Andersen foi a primeira patologia descrita na seção sobre canalopatias - patologias de canais iônicos. A síndrome de Andersen é herdada de forma autossômica dominante, embora haja relatos de casos esporádicos. A probabilidade de transmissão por herança é superior a 50%. A gravidade da doença pode variar dentro de uma família - uma criança pode apresentar lesões graves, enquanto outra pode ser clinicamente assintomática. A penetrância da doença é amplamente variável e nem todos os pacientes apresentam toda a gama de características clínicas desta síndrome. Os distúrbios do ritmo acompanham qualquer ataque de paralisia muscular, ocorrendo secundariamente e causados por flutuações bruscas no nível de potássio no soro sanguíneo do paciente (fig. 1). Porém, a literatura descreve casos em que os distúrbios do ritmo foram a primeira manifestação clínica da síndrome de Andersen e precederam episódios de paresia e paralisia muscular. Neste fenótipo da doença, foi posteriormente identificado um intervalo QT prolongado. Casos de morte cardíaca súbita também foram relatados na síndrome de Andersen.
Anais de arritmologia, nº 4, 2005
Anais de arritmologia, nº 4, 2005
III FAV V3 * V6 Soro K+=2,9
III FAV V3 V6 Soro K+=3,4
Arroz. 1. Um episódio de bigeminismo ventricular em uma menina de 16 anos com síndrome de Andersen num contexto de hipocalemia (a) (os asteriscos indicam extra-sístoles ventriculares); b - a normalização do nível de potássio no sangue levou ao desaparecimento da arritmia.
K+ sérico - potássio sérico.
Apesar de sua baixa prevalência na população, a síndrome de Andersen é de grande interesse científico e prático para a medicina moderna, pois é a única entre todas as canalopatias geneticamente determinadas que afeta tanto os músculos estriados (esqueléticos) quanto o músculo cardíaco. Outras doenças que se manifestam como paresia e paralisia muscular são causadas por mutações em genes responsáveis pelo transporte de íons sódio, cálcio e potássio nas células musculares esqueléticas, enquanto várias formas de síndrome do QT longo são causadas por mutações em genes que codificam o transporte de sódio e íons de potássio apenas em cardiomiócitos. Estudos anteriores excluíram a possibilidade de gênese alélica da síndrome de Andersen. No entanto, em 2001, N. Plaster et al. Ao realizar um estudo genético molecular em uma família grande (15 indivíduos), descobrimos uma conexão entre a probabilidade de herdar a síndrome de Andersen e a patologia do locus 17q do cromossomo 23 e identificamos uma mutação heterozigótica missense do gene KCNJ2 (o locus 17q do cromossomo 23 cruza com o locus do gene KCNJ2 - 17q23.1-17q24 .2). Mutações do gene KCNJ2 foram encontradas em mais de 50% dos pacientes com síndrome de Andersen, confirmando assim o fato de que o gene KCNJ2 é responsável pelo desenvolvimento desta patologia. Atualmente, foram identificadas mais de 20 mutações heterozigóticas missense do gene KCNJ2 que causam a síndrome de Andersen (Fig. 2). Os genes da família KCNJ são amplamente expressos em vários tecidos: músculo (KCNJ2, KCNJ11), coração (KCNJ2, KCNJ3, KCNJ5, KCNJ11), cérebro (KCNJ3, KCNJ6, KCNJ9, KCNJ11), epitélio (KCNJ1, KCNJ2) e muitos outros. Mutações na família de genes KCNJ podem levar ao desenvolvimento de três doenças hereditárias em humanos e uma doença em camundongos. Assim, mutações no gene KCNJ1 (Kir1.1) levam ao desenvolvimento da síndrome de Bart-ter, uma doença autossômica recessiva caracterizada por hipocalemia e perda de sódio no corpo humano; mutações do gene KCNJ11 (Kir6.2) e da proteína associada SUR1 levam ao desenvolvimento de hipoglicemia hiperinsulinêmica permanente.
Arroz. 2. Estrutura da subunidade do canal Kir2.1.
Poro – furo central (tempo); M1, M2 - domínios transmembrana; extracelular - extracelular; intracelular - intracelular.
As caixas cinzentas indicam as 20 mutações atualmente identificadas em pacientes com síndrome de Andersen; o quadrado branco indica a mutação identificada na síndrome do QT curto.
glicemia em crianças, mutações Kir3.2 (GIRK2) - a uma patologia autossômica recessiva em camundongos, manifestada por perda de neurônios e ataxia grave e, finalmente, mutações no gene KCNJ2 estão associadas ao desenvolvimento da síndrome de Andersen. KCNJ2 codifica a síntese da proteína Kir2.1, que faz parte do canal de potássio em células capazes de excitabilidade, incluindo cardiomiócitos, células de músculo estriado e cérebro. Os canais Kir2.1 são os reguladores mais importantes do potencial de membrana em repouso dos músculos cardíacos e esqueléticos e, portanto, da excitabilidade das células desses tecidos em geral, pois garantem a liberação de íons potássio das células com membrana hiperpolarizada durante o final fase de repolarização do potencial de ação. A proteína Kir2.1 consiste em 427 aminoácidos com dois domínios transmembrana (M1, M2) e um orifício central (poro) (H5) e regula o componente IK1 da corrente de potássio com retificação retardada da fase de repolarização (ver Fig. 2 ). A análise Northern blot revelou uma transcrição de 5,5 kb do gene KCNJ2 com altos níveis de Kir2.1 no coração, cérebro, placenta, pulmões e músculo esquelético e níveis mais baixos nos rins. No coração, os canais Kir2.1 predominam nos átrios, ventrículos (com alta velocidade de condução da corrente IK1) e fibras de Purkinje e são menos comuns nas células nodais. As subunidades Kir2.1, codificadas pelo gene KCNJ2, combinam-se em um tetrâmero dentro da parede celular dos cardiomiócitos, formando um canal funcional; eles também podem combinar-se com outras subunidades da família Kir2.1 como heteromultímeros, indicando sua complexidade e diversidade funcional. A maioria das mutações do gene KCNJ2 são mutações missense.
tendo um efeito negativo dominante, o que leva à diminuição da corrente IK1, à diminuição da repolarização e ao aumento da duração do potencial de ação. Como resultado dos processos acima, ocorre despolarização e desestabilização do potencial de membrana em repouso, o que inicia o aparecimento de arritmias ventriculares ou contrações miotônicas dos músculos esqueléticos. Além disso, os pesquisadores sugerem que a disfunção do canal Kir2.1 e a diminuição da corrente IK1 desempenham um papel importante no desenvolvimento de disfunções de sinalização em tecidos não excitáveis, o que pode explicar as manifestações de dismorfogênese em pacientes com síndrome de Andersen. M. Tristani-Firouzi et al. estudaram as consequências funcionais das mutações na síndrome de Andersen, expressando a proteína alterada in vitro. Comprometimento significativo da função do canal Kir2.1 foi observado em todos os tipos de mutações identificadas até o momento.
As manifestações clínicas da síndrome de Andersen são extremamente variáveis, o que se explica pela existência de diversas mutações que contribuem para a manifestação de diferentes fenótipos da doença. É provável que defeitos em outros canais iônicos também sejam importantes na etiologia da síndrome de Andersen. A paralisia muscular recorrente pode ser causada por patologia dos canais de cálcio, sódio e potássio dos miócitos. Os pacientes apresentam episódios de curta duração de fraqueza nos braços e pernas, até paresia generalizada e paralisia dos membros. Esses ataques podem ocorrer em repouso, após o exercício ou ao acordar pela manhã. Ressalta-se que os fatores desencadeantes da doença são diferentes. Atualmente, nem todos os fatores nutricionais que provocam um ataque de paralisia muscular na síndrome de Andersen são conhecidos. É provável que os gatilhos incluam alimentos ricos em potássio e glicose. Outros fatores que provocam a ocorrência de paralisia muscular periódica podem ser: mudança nas atividades - descanso após atividade física, atividade física após um longo período sentado, acordar após dormir, uma longa caminhada com o estômago vazio, uma grande refeição. Qualquer resfriado também pode causar paralisia muscular; Há relatos na literatura de casos em que os gatilhos foram gases inalados – dióxido de carbono, vapores de gasolina ou mesmo cheiro de tinta a óleo. No estudo Tristani-Firuozi, foi observada paralisia muscular cíclica em 23 (64%) dos 36 pacientes com mutações no gene KCNJ2. O repouso após a atividade física foi o gatilho mais comum para a ocorrência de paralisia muscular, como nas variantes clássicas desse tipo.
condições, com a maioria (55%) dos pacientes apresentando hipocalemia (concentração sérica de potássio menor ou igual a 3,4 mEq/L), hipercalemia foi observada em 22% e normocalemia em 10% dos pacientes. Porém, na síndrome de Andersen, os quadros hipocalêmicos não foram precedidos pela ingestão de carboidratos, como é o caso das variantes clássicas da paralisia muscular hipocalêmica. A biópsia muscular realizada em 12 pacientes revelou alterações inespecíficas - pequenas miopatias e/ou agregados tubulares observados em outras formas de paresia ou paralisia muscular cíclica. Os inibidores da anidrase de carbono foram eficazes na redução da frequência de ataques de paralisia na síndrome de Andersen, bem como nas formas clássicas de paralisia muscular.
As alterações mais características no ECG em pacientes com síndrome de Andersen são prolongamento do intervalo OT e ondas de alta amplitude e (Fig. 3). Atualmente, existem opiniões conflitantes entre os pesquisadores quanto ao envolvimento da síndrome de Andersen em uma das variantes da síndrome do intervalo OT longo. Por ser considerada uma patologia geneticamente determinada de repolarização das células musculares, a síndrome de Andersen passou a ser classificada como síndrome do intervalo OT longo (LTS), nomeadamente tipo 7 (LOTS). Atualmente, foram identificados 5 genes, cujas mutações são confiáveis responsáveis pelo desenvolvimento de manifestações clínicas típicas da síndrome do intervalo OT longo: KSYO1 (SHT1), IEAO (KSYN2, SHT2), SSY5L (SHT3), KSYO1 (SHT5), KSYO2 (SHT6). Semelhante a outras formas de síndrome congênita de OT prolongada, a manifestação primária é
Arroz. 3. As alterações eletrocardiográficas e distúrbios do ritmo mais típicos detectados em pacientes com síndrome de Andersen. a - prolongamento do intervalo OT; b - taquicardia ventricular polimórfica instável de curta duração seguida de bigeminismo ventricular; c - taquicardia ventricular bidirecional; d - as setas indicam dentes salientes e.
Anais de arritmologia, nº 4, 2005
Anais de arritmologia, nº 4, 2005
A principal característica do sistema cardiovascular na síndrome de Andersen foi o prolongamento do intervalo OT, identificado em 71% de todos os portadores do gene KSH2. O valor médio do intervalo TO corrigido (OTI) em probandos masculinos e femininos com síndrome de Andersen foi de 479 e 493 ms, respectivamente, enquanto em outras formas de síndrome de TO longo foi de 497 e 510 ms, respectivamente. Devido à presença de um intervalo de OT prolongado e à possibilidade de desenvolver arritmias ventriculares com risco de vida, alguns pesquisadores consideram a síndrome de Andersen como um dos subtipos da síndrome de OT congênita prolongada. No entanto, como se viu, apesar da ocorrência frequente de arritmias ventriculares (extrassístoles ventriculares e séries de taquicardias ventriculares não sustentadas, incluindo taquicardias ventriculares bidirecionais, ver Fig. 3) - em 64% dos pacientes no estudo M. Il81ash-R1goi21, o o risco de mortalidade cardíaca súbita na síndrome de Andersen foi menor do que na síndrome do intervalo OT longo e outras canalopatias hereditárias. Para identificar os mecanismos de ocorrência de arritmias ventriculares na síndrome de Andersen, os autores simularam em um experimento a supressão da função do canal Yu2.1 dos cardiomiócitos do ventrículo do coração. Descobriu-se que a supressão da função do canal Yu2.1 no contexto da hipocalemia levou a um prolongamento da fase final do potencial de ação do músculo cardíaco, à ocorrência de pós-despolarizações retardadas devido à indução da troca Ca+/Ca2+ e à desenvolvimento de arritmias espontâneas. Os autores concluíram
que o substrato para a ocorrência de arritmias ventriculares na síndrome de Andersen difere daquele de outras formas de síndrome congênita do intervalo OT longo e está mais próximo das arritmias resultantes de sobrecarga de Ca2+ ou intoxicação digitálica (como taquicardia ventricular bidirecional e polimórfica). Assim, na síndrome de Andersen, as manifestações clínicas da síndrome congênita do intervalo OT longo e da taquicardia ventricular polimórfica familiar (catecolaminérgica) são combinadas (com esta última, via de regra, não se observa intervalo OT prolongado). A gama de distúrbios do ritmo encontrados na síndrome de Andersen é apresentada na tabela.
A alta amplitude das ondas é um achado específico na síndrome de Andersen e é registrada principalmente nas derivações torácicas anteriores (ver Fig. 3). Esta característica foi detectada em 76% dos probandos e 47% dos portadores de genes KSH2 mutantes de acordo com M. ln81at-Igou21. Deve-se notar que normalmente em pessoas saudáveis a onda pode ser registrada em uma frequência cardíaca rara, enquanto em probandos com síndrome de Andersen no estudo de M. ln81at-Igoi21 a frequência cardíaca média foi de 84±17 batimentos/min (de 52 a 115 batimentos/min).min). Sabe-se que na hipocalemia a amplitude da onda também pode ser elevada, porém não encontramos estudos relatando o nível de potássio no soro sanguíneo em pacientes com ondas de alta amplitude, portanto o papel da hipocalemia na gênese de ondas de alta amplitude e na síndrome de Andersen não podem ser excluídas. Auto-
Manifestações clínicas da síndrome de Andersen em probandos com o gene KSP mutante
Grupo de mutações Gênero Frequência cardíaca Duração OT, ms Distúrbios do ritmo
D71V 4415 F 100 513 Não detectado
A95-98 (supressão A) 3328 F 83 475 Bigeminismo, VT polimórfico
8136B 6634 F 68 500 Bigeminia
P186b 7246 M 94 PBPNPG* VT Polimórfico
R218W 2401 F 100 560 Bigeminismo, parada cardíaca não fatal, bloqueio AV de 1º grau, fibrilação, flutter ventricular
R218W 2679 M 68 510 Bigeminia, VT polimórfico
R218W 2681 F 75 488 Bigeminy, VT polimórfico
R218W 7480 M 94 525 Bigeminia
R218W 6515 M 52 416 Bigeminia, VT polimórfico
R218Q 6562 M 70 469 Não detectado
G300V 3677 F 100 480 Bigeminia, VT polimórfico
V302M 2682 M 79 PBPNPG* Bigeminia
E303K 2281 F 85 495 Extrassístoles ventriculares frequentes
A314-315 5768 F 115 471 Fibrilação, flutter ventricular, TV monomórfica
* O bloqueio completo do ramo direito impediu a medição precisa do intervalo OT.
Os pesquisadores também avaliaram a relação entre a gravidade das mutações (gravidade da disfunção do canal Kr2.1) e a gravidade das manifestações clínicas (prolongamento do TPV, arritmias, sintomas neuromusculares, dismorfogênese) da síndrome de Andersen. Descobriu-se que o fenótipo clínico da doença não se correlacionou com o grau de supressão dominante negativa da função do canal K1r2.1.
G. Andelfinge et al. identificaram a mutação missense heterozigótica R67W do gene KSH2 em 41 parentes consangüíneos e descobriu-se que a herança de arritmias ventriculares e paralisia muscular periódica diferia por sexo: assim, as arritmias ventriculares predominaram em mulheres (13 de 16, ou 81%) e sintomas neuromusculares - em homens (10 em 25, ou 40%). Ao mesmo tempo, as arritmias ventriculares começaram a aparecer após os 10 anos de idade e eram menos frequentes durante a gravidez (enquanto com outros tipos de mutações do gene KSH2 em mulheres, os distúrbios do ritmo eram mais frequentes durante a gravidez) e após 55 anos (durante menopausa). Notavelmente, não houve prolongamento do intervalo OT neste pedigree. Neste grupo de pacientes, três apresentavam história de síncope; Um paciente que sobreviveu à morte cardíaca súbita recebeu um cardioversor desfibrilador implantado. Em 8 pacientes do sexo masculino, foram observados episódios de fraqueza muscular ou paralisia após o exercício e em 2 houve hipocalemia. Hipertelorismo foi observado em 4 indivíduos, mandíbula pequena em 10, sindactilia de dedos das mãos ou dos pés em 9, clinodactilia em 12. Sinais de dismorfogênese foram igualmente comuns em homens e mulheres. Um paciente do sexo masculino foi submetido a tratamento cirúrgico para escoliose e uma paciente do sexo feminino para fissura de palato. Muito incomum foi a identificação no pedigree estudado de displasia renal unilateral e patologia valvar do coração - estenose valvar pulmonar (diagnosticada em um paciente aos 6 meses de idade), valva aórtica bicúspide (em 3 pacientes) e valva aórtica bicúspide com coarctação da aorta (em 1 paciente). Tais anormalidades foram identificadas pela primeira vez em pacientes com síndrome de Andersen. Além disso, houve relato de morte de um recém-nascido com cardiopatia desconhecida. Bloqueio atrioventricular (AV) de primeiro grau foi observado em 4 homens e uma mulher em combinação com bloqueio de ramo esquerdo. Nenhum indivíduo apresentou todas as manifestações características da síndrome de Andersen ao mesmo tempo. Pleiotropia das manifestações fenotípicas na síndrome de Andersen dentro
Esse pedigree (Fig. 4) pode ser explicado tanto pelo efeito específico da mutação R67W, quanto pela variação na expressão dos alelos, ou pelo efeito modificador de fatores externos.
Existem poucas descrições de tratamentos utilizados para a síndrome de Andersen. Como exemplo, citamos o relatório de J. Junker et al. : em paciente de 6 anos sem história familiar de doenças hereditárias ou cardiovasculares, começaram a ocorrer pela primeira vez episódios recorrentes de paresia atônica. Aos 10 anos de idade, ela foi suspeita de miocardite pós-estreptocócica devido a um alto nível de creatinase sérica (até 447 U/L) e extra-sístoles ventriculares polimórficas (PVCs) assintomáticas registradas no ECG. Devido à interrupção espontânea dos ataques de fraqueza muscular, os médicos não excluíram a possibilidade de sua origem psicogênica. Aos 15 anos, o paciente apresentou episódio de fibrilação ventricular (FV) e a reanimação cardiopulmonar foi realizada com sucesso. A estimulação elétrica programada induziu taquicardia ventricular sustentada, com QTc de 0,45 s. O paciente foi implantado com um cardioversor-desfibrilador (CDI). Após um ano, começaram a ocorrer ataques diários de FV, necessitando de choques com CDI, e foram observados episódios de fraqueza muscular grave (até mesmo paralisia muscular). Entre as características que chamaram a atenção durante o exame físico estavam: base larga do nariz, clinodactilia, escoliose e baixa estatura. A paralisia muscular atônica cobriu diferentes grupos musculares das extremidades superiores e inferiores, ocorreu repentinamente, durou várias horas ou dias e parou por conta própria. Os níveis séricos de potássio e creatina quinase estavam normais. Os ataques de fraqueza foram induzidos por hipercalemia ou frio, e o exercício foi acompanhado por uma diminuição do potencial de ação muscular. A biópsia muscular revelou agregados tubulares e vários vacúolos. O diagnóstico clínico da variante esporádica da síndrome de Andersen foi confirmado após um estudo de genética molecular que revelou uma mutação missense heterozigótica R218W do gene KCNJ2 na menina, mas não em seus pais. O sotalol e os antiarrítmicos classe I (AAAs), incluindo flecainida e propafenona, foram ineficazes. A amiodarona foi adicionada à terapia com captopril, nadolol e digitoxina em dose de ataque de 200 mg por dia. As arritmias ventriculares desapareceram rapidamente e os ataques de fraqueza muscular continuaram a ocorrer. Após 2 meses, acetazolamida (Diamox) foi adicionada à terapia acima na dose de 750 mg por dia. Nos próximos 2 anos, o paciente
Anais de arritmologia, nº 4, 2005
Anais de arritmologia, nº 4, 2005
1 relatam a falha da terapia com amiodarona e acetazolamida em pacientes com síndrome de Andersen ou a necessidade de interrompê-la devido ao desenvolvimento de efeitos colaterais. Por outro lado, pode haver interação farmacogenética entre a mutação R218W e a resposta terapêutica à amiodarona e acetazolamida, o que requer confirmação em outros pacientes. Como a amiodarona inibe a corrente IK1, é lógico supor que ela inibe a hiperexcitabilidade do músculo cardíaco ao retardar a função dos canais de sódio e cálcio, bem como dos receptores beta-adrenérgicos.
vala, corrigindo assim as alterações causadas pela perda de função do canal Sul2.1. Contudo, devido ao potencial de efeitos colaterais significativos, a amiodarona pode ser recomendada em pacientes com arritmias sintomáticas. A acetazolamida, medicamento que altera a acidez do sangue, pode prevenir crises de paralisia muscular periódica de qualquer origem devido à abertura seletiva dos canais Ksa2+ musculares, o que pode compensar a disfunção da corrente IK1 e evitar queda no potencial de membrana dos músculos na síndrome de Andersen. Pacientes com formas hipocalêmicas de paresia muscular podem tomar cloreto de potássio dissolvido em uma solução sem açúcar (os sintomas geralmente desaparecem em uma hora); eles devem evitar alimentos ricos em carboidratos e exercícios excessivos. Pacientes com formas hipercalêmicas de paralisia muscular podem prevenir esses ataques comendo refeições frequentes, ricas em carboidratos e com baixo teor de potássio.
Assim, apresentamos a revisão mais abrangente até o momento sobre o quadro clínico, diagnóstico e tratamento da síndrome de Andersen, uma das patologias únicas entre as canalopatias hereditárias devido à combinação incomum de manifestações fenotípicas dos sistemas cardíaco e músculo-esquelético em combinação com vários
sinais figurativos de dismorfogênese, cujo mecanismo de ocorrência ainda permanece obscuro. Semelhante à descoberta de novas variantes de doenças geneticamente determinadas, como a síndrome do QT longo ou curto, novas pesquisas no campo da genética molecular ajudarão a identificar novas formas da síndrome de Andersen e a estudar mais profundamente os mecanismos de ação das mutações (as mutações podem perturbar o funções dos canais iônicos e proteínas transmembrana em diferentes níveis de sua interação) e explicam as manifestações fenotípicas incomuns desta doença, e estudos farmacogenéticos ajudarão a desenvolver novos métodos de tratamento.
LITERATURA
1. Andersen E. D., Krasilnikoff P. A., Overad H. Fraqueza muscular intermitente, extrassístoles e múltiplas anormalidades de desenvolvimento: Uma nova síndrome? // Acta Pediatr. Escândalo.
1971. -Vol. 60. - S. 559-564.
2. Andelfinger G., Tapper A.R., Welch R.C. et al. A mutação KCNJ2 resulta na síndrome de Andersen com fenótipos musculares cardíacos e esqueléticos específicos do sexo // Amer. J.Hum. Geneta. - 2002. - Vol. 71. - S. 663-668.
3. BakerN., Iannaccone ST, BurnsD., sincronia de Scott W. Andersen
drome: Fraqueza episódica e disritmia ventricular familiar // J. Child. Neurol. - 1996. - Vl. 11. - P. 152 (Resumo).
4. Bendahhou S., Donaldson MR, Plaster NM et al. Defec-
O tráfego ativo do canal de potássio Kir2.1 está subjacente à síndrome de Andersen-Tawil // J. Biol. Química. - 2003. - Vol. 278, nº 51. - S. 51779-51785.
5. Bosch R. F., Li G. R., Gaspo R., Nattel S. Efeitos eletrofisiológicos da terapia crônica com amiodarona e hipotireoidismo, isoladamente e em combinação, em miócitos ventriculares de cobaia // J. Pharmacol. Exp. Lá. - 1999. - Vol. 289. - S. 156-165.
6. Canun S., PerezN., Beirana L.G. Síndrome de Andersen autossômica dominante em três gerações // Amer. J. Med. Geneta. - 1999. - Vol. 85. - S. 147-156.
7. Jen J., Ptacek L. J. Canalopatias // Bases metabólicas e moleculares de doenças hereditárias / C. R. Scriver, A. L. Beaudet, W. S. Sly, D. Valle (eds). - NY: McGraw-Hill, 2001.- P. 5223-5238.
ne e acetazolamida para o tratamento da síndrome de Andersen grave geneticamente confirmada // Neurologia. - 2002.
Vol. 59, nº 3. - P. 466.
9. Lange PS, Er F., Gassanov N., Hoppe UC Andersen
mutações de KCNJ2 suprimem o retificador interno nativo
atual IK1 de forma dominante negativa // Cardiovasc. Res. - 2003. - Vol. 59, nº 2. - S. 321-327.
10. Kimbrough J. et al. Implicações clínicas para pais e irmãos afetados de probandos com síndrome do QT longo // Circulação. - 2001. - Vol. 104. - S. 557-562.
11. Klein R, Genelin R., Marks J. F. Paralisia periódica com arritmia cardíaca // J. Pediatr. - 1965. - Vol. 62. - S. 371-385.
12. Keating M. T., Sanguinetti M. C. Mecanismos moleculares e celulares de arritmias cardíacas // Célula. - 2001. - Vol. 104.
13. Levitt L. P., Rose L. I., Dawson D. M. Paralisia periódica hipocalêmica com arritmia // N. Engl. J. Med. - 1972.
Vol. 286. - S. 253-254.
14. Gesso N. M. et al. Mutações em Kir2.1 causam os fenótipos elétricos episódicos e de desenvolvimento da síndrome de Andersen // Cell. - 2001. - Vol. 105. - S. 511-519.
15. Pouget J., Philip N., Faugere G., Pellissier J. F. Síndrome de Andersen: Uma forma particular de paralisia com disritmia cardíaca // Rev. Neurol. (Paris). - 2004. - Vol. 160, nº 5 (parte 2). - S. 38-42 (francês).
16. Sansone Ket al. Síndrome de Andersen: Uma paralisia periódica distinta // Ann. Neurol. - 1997. - Vol. 42. - S. 305-312.
17. Schulze-Bahr E. Síndrome do QT curto ou síndrome de Andersen: Yin e yang da disfunção do canal Kir2.1 // Circ. Res. - 2005. - Vol. 96. - S. 703-704.
18. Onda Surawicz B. U: Fatos, hipóteses, equívocos e nomes incorretos // J. Cardiovasc. Eletrofisiol. - 1998.
Vol. 9. - S. 1117-1128.
19. Tawil R. et al. Síndrome de Andersen: paralisia periódica sensível ao potássio, ectopia ventricular e características dismórficas // Ann. Neurol. - 1994. - Vol. 35. - S. 326-330.
20. Tawil R. et al. Ensaios randomizados de diclorfenamida nas paralisias periódicas. Grupo de Trabalho sobre Paralisia Periódica // Ibid. - 2000. - Vol. 47. - P. 46-53.
21. Tricarico D., Barbieri M., Conte Camerino D. Acetazolamida abre o canal KCa2+ muscular: Um novo mecanismo de ação que pode explicar o efeito terapêutico da droga na paralisia periódica hipocalêmica // Ibid. - 2000. - Vol. 48.
22. Tristani-Firouzi M., Jensen JL, Donaldson MR et al. Caracterização funcional e clínica de mutações KCNJ2 associadas a LQT7 (síndrome de Andersen) // J. Clin. Investir. - 2002. - Vol. 110, nº 3. - S. 381-388.
UDC 616.124.3:616.127]-07-08
DIAGNÓSTICO, CURSO E TRATAMENTO DA CARDIOMIOPATIA/DISPLASIA ARRITMMOGÊNICA AUTOSÔMICA DOMINANTE DO VENTRÍCULO DIREITO
LA Bockeria, VA Bazaev, A. Kh. Melikulov, UT Kabaev, OL Bockeria, RV Viskov,
A G. Filatov, A N. Gritsai, V. V. Chumakov
Centro Científico de Cirurgia Cardiovascular em homenagem. A. N. Bakuleva (diretor - Acadêmico da Academia Russa de Ciências Médicas L. A. Bockeria) RAMS, Moscou
displasia ritmogênica do ventrículo direito ou cardiomiopatia/displasia arritmogênica do ventrículo direito - uma patologia de etiologia desconhecida, muitas vezes hereditária, caracterizada por risco de vida
infiltração opaca ou fibrogordurosa do miocárdio, predominantemente no pâncreas, acompanhada por arritmias ventriculares de gravidade variável, incluindo fibrilação ventricular.
Anais de arritmologia, nº 4, 2005
Glicogenose tipo IV (doença de Andersen, amilopectinose, glicogenose difusa com cirrose hepática)- uma doença hereditária causada por uma deficiência de enzimas envolvidas no metabolismo do glicogénio; caracterizada por uma violação da estrutura do glicogênio, seu acúmulo insuficiente ou excessivo em vários órgãos e tecidos.
Doença de Andersen ocorre como resultado de mutações no gene microssomal da amilo-1,4:1,6-glucano transferase, levando à sua deficiência no fígado, músculos, leucócitos, eritrócitos e fibroblastos. O gene está mapeado no cromossomo 3p 12. O modo de herança é autossômico recessivo.
A amilo-1,4:1,6-glucano transferase está envolvida na síntese de glicogênio em pontos de ramificação da árvore do glicogênio. A enzima conecta uma sessão de pelo menos seis resíduos glicosídicos ligados a α-1,4 das cadeias externas de glicogênio à “árvore” de glicogênio por meio de uma ligação α-1,6-glicosídica. Quando a enzima é deficiente, a amilopectina é depositada nas células do fígado e dos músculos, o que leva a danos celulares. A concentração de glicogênio no fígado não excede 5%.
Doença manifesta-se no primeiro ano de vida com sintomas gastrointestinais inespecíficos: vômitos, diarreia. À medida que a doença progride, ocorrem hepatoesplenomegalia, insuficiência hepática progressiva, hipotonia e atrofia muscular generalizada e cardiomiopatia grave. A morte dos pacientes geralmente ocorre antes dos 3-5 anos de idade devido à insuficiência hepática crônica, raramente na infância (até 8 anos).
Diagnóstico laboratorial com base na detecção de glicogênio com estrutura alterada em biópsia hepática e diminuição da atividade da amilo-1,4:1,6-glucano transferase.
O tratamento visa combater distúrbios metabólicos, incl. com acidose. Em alguns casos, o uso de glucagon, hormônios anabólicos e glicocorticóides é eficaz. Refeições frequentes ricas em carboidratos de fácil digestão são necessárias para a hipoglicemia. Nas formas musculares de glicogenose, a melhora é observada seguindo uma dieta rica em proteínas, administrando frutose (50-100 g por via oral por dia), multivitaminas e ATP. Estão sendo feitas tentativas de administrar enzimas ausentes aos pacientes.
Os pacientes com glicogenose estão sujeitos à observação do dispensário por um médico do centro médico de genética e por um pediatra (terapeuta) da clínica.
A prevenção não foi desenvolvida. Para prevenir o nascimento de uma criança com glicogenose em famílias onde havia pacientes semelhantes, é realizado aconselhamento médico e genético.
A doença de Andersen (glicogenose tipo IV, amilopectinose) ocorre devido à deficiência da enzima ramificada 1,4-a-glucano, que leva ao acúmulo de glicogênio anormal e pouco solúvel.
Esta doença é chamada de amilopectinose porque o glicogênio nesses casos é menos ramificado e possui seções lineares mais longas contendo ligações α-1,4-glicosídicas, o que é característico da estrutura da amilopectina.
A doença de Andersen é herdada de forma autossômica recessiva. O gene da enzima ramificada 1,4-a-glucano está localizado no cromossomo 3; Suas mutações subjacentes à doença são conhecidas e suas características em cada caso permitem prever o quadro clínico da doença.
Sintomas da doença de Andersen
A amilopectinose é clinicamente heterogênea. Sua forma clássica mais comum é caracterizada por cirrose hepática progressiva. Os sinais iniciais – hepatoesplenomegalia e mau desenvolvimento – aparecem nos primeiros 18 meses. vida. Hipertensão portal, ascite, veias varicosas do esôfago e insuficiência hepática se desenvolvem gradualmente, das quais os pacientes morrem aos 5 anos de idade. Em casos raros, o dano hepático não progride.
Também há relatos de uma forma neuromuscular da doença de Andersen. Suas manifestações são variadas:
dano difuso ao sistema nervoso periférico central, acompanhado pelo acúmulo de corpos poliglucosanos nos neurônios (a chamada doença dos corpos poliglucosanos). O diagnóstico neste último caso requer a determinação da atividade da ramificação 1,4-a-glucano enzima em leucócitos ou biópsias de tecido nervoso, pois sua deficiência é limitada justamente a essas células.
Diagnóstico da doença de Andersen
A deposição de glicogênio atípico é encontrada no fígado, coração, músculos, pele, intestinos, cérebro e medula espinhal e nervos periféricos. A cirrose nodular pequena se desenvolve no fígado. Quando examinados, inclusões basofílicas fracamente coradas são visíveis nos hepatócitos, que são depósitos PAS-positivos de granulação grossa, parcialmente resistentes à amilase. A microscopia eletrônica revela, além de partículas de glicogênio, agregados fibrosos característicos da amilopectina. A coloração característica das inclusões citoplasmáticas e a imagem microscópica eletrônica poderiam ter valor diagnóstico, mas características histológicas semelhantes foram observadas em polissacaridoses sem deficiência da enzima ramificadora 1,4-a-glucana. Para confirmar o diagnóstico, é necessário estabelecer uma deficiência desta enzima específica no fígado, músculos, cultura de fibroblastos da pele ou leucócitos. Para efeitos de diagnóstico pré-natal, a actividade da enzima ramificada 1,4-a-glucano é determinada em cultura de amniócitos ou vilosidades coriónicas.

Em 2018, o Jejum da Natividade começará em 28 de novembro. Durante este período, os crentes ortodoxos se preparam para celebrar o Natal...

Constituir família é o sonho da maioria das mulheres. Eles querem ter um marido amoroso e um monte de filhos. Mas nem sempre é um relacionamento...

Este artigo contém: a oração mais poderosa pelo divórcio - informações retiradas de todo o mundo, da rede eletrônica e...

Site de informações sobre ícones, orações, tradições ortodoxas. Oração pelos escândalos e brigas na família, com o marido, com os filhos...

O que seria do Ano Novo sem champanhe, tangerinas, Olivier, formol e o “arenque com casaco de pele” favorito de todos. Com o último...

Vamos preparar os ingredientes necessários para os biscoitos. A primeira coisa a fazer é colocar a água para ferver. Nós precisamos...

É possível cadastrar funcionário para o cargo de diretor financeiro - contador-chefe? O contador-chefe afirma...

O chefe de uma pequena empresa pode gerenciar facilmente o orçamento de forma independente. VERIFICADO! Se você conseguir...

A criação de novos projetos envolve uma justificação económica preliminar para a sua viabilidade, posterior...
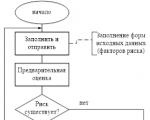
O reporte é gerado pelo RM, é acordado (aprovado) pelo Comité de Risco no âmbito do Conselho de Administração e transmitido a...

Na beira de um prado grande, muito grande, em uma longa folha de grama esmeralda vivia uma pequena joaninha. O pequeno de Deus...

Hoje em dia é bastante comum as pessoas se voltarem para as estrelas. Com a ajuda de um horóscopo uma pessoa pode descobrir...

Negócios ou amigável. Se você estava perseguindo um estranho, isso significa o seu nível de confiança no sexo feminino...

de acordo com o livro dos sonhos de Freud Se você sonhou que estava pescando, isso significa que na vida real dificilmente você consegue desligar...

Constituir família é o sonho da maioria das mulheres. Eles querem ter um marido amoroso e um monte de filhos. Mas nem sempre é...

Este artigo contém: a oração mais poderosa pelo divórcio - informações retiradas de todo o mundo, eletrônicas...