O que é possível e o que não é possível durante o Jejum da Natividade?
Em 2018, o Jejum da Natividade começará em 28 de novembro. Durante este período, os crentes ortodoxos se preparam para celebrar o Natal...
solução de substância: pacotes
Reg. Nº: LSR-007009/08substância -solução.
garrafas (1) - embalagens de papelão.
Interferon. É uma proteína recombinante altamente purificada com peso molecular de 19.300 daltons. Obtido a partir de um clone de Escherichia coli por hibridização de plasmídeos bacterianos com o gene de leucócitos humanos que codifica a síntese de interferon. Ao contrário do interferon, o alfa-2a possui arginina na posição 23.
Tem efeito antiviral, que se deve à interação com receptores de membrana específicos e à indução da síntese de RNA e, em última instância, de proteínas. Estes últimos, por sua vez, impedem a reprodução normal do vírus ou a sua libertação.
Possui atividade imunomoduladora, que está associada à ativação da fagocitose, estimulação da formação de anticorpos e linfocinas.
Tem efeito antiproliferativo nas células tumorais.
Hepatite B aguda, hepatite B crônica, hepatite C crônica.
Leucemia de células pilosas, leucemia mieloide crônica, carcinoma de células renais, sarcoma de Kaposi devido à AIDS, linfoma cutâneo de células T (micose fungóide e síndrome de Sézary), melanoma maligno.
Administrado por via intravenosa ou subcutânea. A dose e o regime de tratamento são definidos individualmente, dependendo das indicações.
Sintomas como os da gripe: frequentemente - febre, calafrios, dores nos ossos, articulações, olhos, mialgia, dor de cabeça, aumento da sudorese, tonturas.
Do sistema digestivo: possível diminuição do apetite, náuseas, vómitos, diarreia, obstipação, alteração do paladar, boca seca, perda de peso, dor abdominal ligeira, ligeiras alterações nos testes de função hepática (geralmente normalizadas após o tratamento).
Do sistema nervoso central e do sistema nervoso periférico: raramente - tontura, deterioração da atividade mental, distúrbios do sono, comprometimento da memória, ansiedade, nervosismo, agressividade, euforia, depressão (após tratamento prolongado), parestesia, neuropatia, tremor; em alguns casos - tendências suicidas, sonolência.
Do sistema cardiovascular: possível - taquicardia (com febre), hipotensão arterial ou hipertensão, arritmia; em alguns casos - distúrbios do sistema cardiovascular, doença arterial coronariana, infarto do miocárdio.
Do sistema respiratório: raramente - dor no peito, tosse, leve falta de ar; em alguns casos - pneumonia, edema pulmonar.
Do sistema hematopoiético: possível leucopenia leve, trombocitopenia, granulocitopenia.
Reações dermatológicas: possível coceira, alopecia reversível.
Outros: raramente - rigidez muscular; em casos isolados - anticorpos contra interferons naturais ou recombinantes.
Doenças cardiovasculares graves, cirrose hepática descompensada, depressão grave, psicose, dependência de álcool ou drogas, aumento da sensibilidade ao interferon alfa-2b.
O uso durante a gravidez só é possível se o benefício esperado da terapia para a mãe superar o risco potencial para o feto.
Não se sabe se o interferon alfa-2b é excretado no leite materno. Caso seja necessário utilizá-lo durante a lactação, deve-se decidir a questão da interrupção da amamentação.
Mulheres em idade fértil devem usar métodos contraceptivos confiáveis durante o tratamento.
Contra-indicado na cirrose hepática descompensada. Use com cautela em pacientes com insuficiência hepática.
Use com cautela em pacientes com insuficiência renal.
Use com cautela em pacientes com insuficiência renal, hepática, hematopoiese da medula óssea ou com tendência a tentativas de suicídio.
Em pacientes com doenças do sistema cardiovascular, é possível arritmia. Se a arritmia não diminuir ou aumentar, a dose deve ser reduzida em 2 vezes ou o tratamento deve ser interrompido.
Durante o período de tratamento, é necessária a monitorização do estado neurológico e mental.
Em casos de supressão grave da hematopoiese da medula óssea, é necessário um exame regular da composição do sangue periférico.
O interferon alfa-2b tem um efeito estimulante no sistema imunológico e deve ser usado com cautela em pacientes propensos a doenças autoimunes devido ao risco aumentado de reações autoimunes.
O interferão alfa-2b inibe o metabolismo da teofilina e reduz a sua depuração.
Sucção
Com administração subcutânea ou intramuscular de interferon alfa-2b, sua biodisponibilidade varia de 80% a 100%. Após a administração de interferon alfa-2b, o Tmax no plasma sanguíneo é de 4-12 horas, T1/2 - 2-6 horas.16-24 horas após a administração, o interferon recombinante não é detectado no soro sanguíneo.
Metabolismo
O metabolismo ocorre no fígado.
Os interferons alfa podem interromper os processos metabólicos oxidativos, reduzindo a atividade das enzimas hepáticas microssomais do sistema citocromo P450.
Remoção
É excretado principalmente pelos rins por filtração glomerular.
Não são fornecidos dados sobre overdose de Altevir®.
O medicamento deve ser armazenado fora do alcance das crianças, conforme SP 3.3.2-1248-03, em temperatura de 2° a 8°C; não congele.
As interações medicamentosas entre Altevir e outros medicamentos não foram totalmente estudadas. Altevir® deve ser usado com cautela simultaneamente com hipnóticos e sedativos, analgésicos narcóticos e medicamentos que potencialmente tenham efeito mielossupressor.
Quando Altevir e teofilina são prescritos simultaneamente, deve-se monitorar a concentração desta no soro sanguíneo e, se necessário, alterar seu regime posológico.
Quando Altevir é utilizado em combinação com medicamentos quimioterápicos (citarabina, ciclofosfamida, doxorrubicina, teniposido), o risco de desenvolver efeitos tóxicos aumenta.
Reações gerais: muitas vezes - febre, fraqueza (são reações dose-dependentes e reversíveis, desaparecem dentro de 72 horas após uma interrupção no tratamento ou sua interrupção), calafrios; menos frequentemente - mal-estar.
Do lado do sistema nervoso central: muitas vezes - dor de cabeça; menos frequentemente - astenia, sonolência, tontura, irritabilidade, insônia, depressão, pensamentos e tentativas de suicídio; raramente - nervosismo, ansiedade.
Do sistema músculo-esquelético: muito frequentemente - mialgia; menos frequentemente - artralgia.
Do sistema digestivo: muitas vezes - perda de apetite, náusea; menos frequentemente - vômito, diarréia, boca seca, alteração do paladar; raramente - dor abdominal, dispepsia; é possível um aumento reversível na atividade das enzimas hepáticas.
Do sistema cardiovascular: muitas vezes - diminuição da pressão arterial; raramente - taquicardia.
Reações dermatológicas: menos frequentemente - alopecia, aumento da sudorese; raramente - erupção cutânea, coceira.
Do sistema hematopoiético: são possíveis leucopenia reversível, granulocitopenia, diminuição dos níveis de hemoglobina e trombocitopenia.
Outros: raramente - perda de peso, tireoidite autoimune.
interferon alfa-2b recombinante humano 3 milhões de UI
Excipientes: acetato de sódio, cloreto de sódio, sal dissódico do ácido etilenodiamino tetraacético, Tween-80, dextrano 40, água para preparações injetáveis.
Aplicar por via subcutânea, intramuscular e intravenosa. O tratamento deve ser iniciado por um médico. Depois, com autorização do médico, o paciente pode administrar uma dose de manutenção de forma independente (nos casos em que o medicamento é prescrito por via subcutânea ou intramuscular).
Hepatite B crônica: Altevir® é administrado por via subcutânea ou intramuscular na dose de 5-10 milhões de UI 3 vezes por semana durante 16-24 semanas. O tratamento é interrompido após 3-4 meses de uso na ausência de dinâmica positiva (de acordo com um estudo do DNA do vírus da hepatite B).
Hepatite C crônica: Altevir® é administrado por via subcutânea ou intramuscular na dose de 3 milhões de UI 3 vezes por semana durante 24-48 semanas. Em pacientes com evolução recidivante da doença e pacientes que não receberam tratamento anterior com interferon alfa-2b, a eficácia do tratamento aumenta com a terapia combinada com ribavirina. A duração da terapia combinada é de pelo menos 24 semanas. A terapia com Altevir deve ser realizada durante 48 semanas em pacientes com hepatite C crônica e 1º genótipo do vírus com carga viral elevada, nos quais o RNA do vírus da hepatite C não é detectado no soro sanguíneo até o final das primeiras 24 semanas de tratamento.
Papilomatose laríngea: Altevir® é administrado por via subcutânea na dose de 3 milhões UI/m2 3 vezes por semana. O tratamento começa após a remoção cirúrgica (ou laser) do tecido tumoral. A dose é selecionada levando-se em consideração a tolerabilidade do medicamento. Alcançar uma resposta positiva pode exigir tratamento durante 6 meses.
Leucemia de células pilosas: a dose recomendada de Altevir para administração subcutânea em pacientes após ou sem esplenectomia é de 2 milhões UI/m2, 3 vezes por semana. Na maioria dos casos, a normalização de um ou mais parâmetros hematológicos ocorre após 1-2 meses de tratamento, sendo possível aumentar o período de tratamento para 6 meses. Este regime posológico deve ser seguido continuamente, a menos que ocorra progressão rápida da doença ou sintomas de intolerância grave ao medicamento.
Leucemia mieloide crónica: a dose recomendada de Altevir em monoterapia é de 4-5 milhões de UI/m2 por dia, por via subcutânea, diariamente. Para manter o número de leucócitos, pode ser necessária uma dose de 0,5-10 milhões UI/m2. Se o tratamento permitir o controle do número de leucócitos, então para manter a remissão hematológica o medicamento deve ser utilizado na dose máxima tolerada (4-10 milhões de UI/m2 diariamente). O medicamento deve ser descontinuado após 8-12 semanas se a terapia não levar à remissão hematológica parcial ou a uma diminuição clinicamente significativa no número de leucócitos.
Linfoma não-Hodgkin: Altevir® é usado como terapia adjuvante em combinação com regimes quimioterápicos padrão. O medicamento é administrado por via subcutânea na dose de 5 milhões de UI/m2, 3 vezes por semana, durante 2 a 3 meses. A dose deve ser ajustada dependendo da tolerabilidade do medicamento.
Melanoma: Altevir® é utilizado como terapia adjuvante quando há alto risco de recidiva em adultos após a remoção do tumor. Altevir® é administrado por via intravenosa na dose de 15 milhões de UI/m2 5 vezes por semana durante 4 semanas, depois por via subcutânea na dose de 10 milhões de UI/m2 3 vezes por semana durante 48 semanas. A dose deve ser ajustada dependendo da tolerabilidade do medicamento.
Mieloma múltiplo: Altevir® é prescrito durante o período de obtenção de remissão estável na dose de 3 milhões de UI/m2 3 vezes por semana por via subcutânea.
Sarcoma de Kaposi devido à AIDS: a dose ideal não foi estabelecida. O medicamento pode ser utilizado em doses de 10 a 12 milhões de UI/m2/dia por via subcutânea ou intramuscular. Se a doença estabilizar ou responder ao tratamento, a terapia é continuada até que ocorra a regressão do tumor ou seja necessária a descontinuação do medicamento.
Câncer renal: a dose e o regime ideais não foram estabelecidos. Recomenda-se o uso do medicamento por via subcutânea em doses de 3 a 10 milhões de UI/m2 3 vezes por semana.
Preparação de solução para administração intravenosa
Extrair o volume de solução de Altevir necessário para preparar a dose necessária, adicionar a 100 ml de solução estéril de cloreto de sódio a 0,9% e administrar durante 20 minutos.
A solução injetável é transparente e incolor.
Use para disfunção hepática
Uso para insuficiência renal
O medicamento é contraindicado em insuficiência renal e/ou hepática grave (incluindo aquelas causadas pela presença de metástases).
Antes do tratamento com Altevir para hepatites virais crônicas B e C, recomenda-se a realização de biópsia hepática para avaliar o grau de dano hepático (sinais de processo inflamatório ativo e/ou fibrose). A eficácia do tratamento da hepatite C crónica aumenta com a terapêutica combinada com Altevir e ribavirina. O uso de Altevir não é eficaz no desenvolvimento de cirrose hepática descompensada ou coma hepático.
Caso ocorram efeitos colaterais durante o tratamento com Altevir, a dose do medicamento deve ser reduzida em 50% ou o medicamento deve ser descontinuado temporariamente até que desapareçam. Se os efeitos secundários persistirem ou recorrerem após a redução da dose, ou se for observada progressão da doença, o tratamento com Altevir deve ser descontinuado.
Se o nível de plaquetas diminuir abaixo de 50x109/l ou o nível de granulócitos abaixo de 0,75x109/l, recomenda-se reduzir a dose de Altevir em 2 vezes com monitorização de análises ao sangue após 1 semana. Se estas alterações persistirem, o medicamento deverá ser descontinuado.
Se o nível de plaquetas diminuir abaixo de 25x109/l ou o nível de granulócitos abaixo de 0,5x109/l, recomenda-se descontinuar Altevir® com monitoramento de exames de sangue após 1 semana.
Em pacientes que recebem preparações de interferon alfa-2b, anticorpos que neutralizam sua atividade antiviral podem ser detectados no soro sanguíneo. Em quase todos os casos, os títulos de anticorpos são baixos e seu aparecimento não leva à diminuição da eficácia do tratamento ou à ocorrência de outras doenças autoimunes.
O medicamento é contraindicado durante a gravidez e lactação (amamentação).
A solução injetável é transparente e incolor.
1 ml
interferon alfa-2b recombinante humano 3 milhões de UI
Excipientes: acetato de sódio, cloreto de sódio, sal dissódico de ácido etilenodiamino tetraacético, Tween-80, dextrano 40, água
18 meses
Como parte da terapia complexa em adultos:
Com hepatite B viral crônica sem sinais de cirrose hepática;
Para hepatite C viral crônica na ausência de sintomas de insuficiência hepática (monoterapia ou terapia combinada com ribavirina);
Com papilomatose da laringe;
Para verrugas genitais;
Para leucemia de células pilosas, leucemia mieloide crônica, linfoma não-Hodgkin, melanoma, mieloma múltiplo, sarcoma de Kaposi devido à AIDS, câncer renal progressivo.
História de doença cardiovascular grave (insuficiência cardíaca crónica não controlada, enfarte do miocárdio recente, perturbações graves do ritmo cardíaco);
Insuficiência renal e/ou hepática grave (incluindo aquelas causadas pela presença de metástases);
Epilepsia, bem como distúrbios graves do sistema nervoso central, especialmente expressos por depressão, pensamentos e tentativas de suicídio (incluindo história);
Hepatite crónica com cirrose hepática descompensada e em doentes que receberam ou receberam recentemente tratamento com imunossupressores (com exceção do tratamento completo de curta duração com corticosteróides);
Hepatite autoimune ou outra doença autoimune;
Tratamento com imunossupressores após transplante;
Doença da glândula tireóide que não pode ser controlada por métodos terapêuticos geralmente aceitos;
Doenças pulmonares descompensadas (incluindo DPOC);
Diabetes mellitus descompensada;
Hipercoagulação (incluindo tromboflebite, embolia pulmonar);
Mielodepressão grave;
Gravidez;
Período de lactação (amamentação);
Hipersensibilidade aos componentes do medicamento.
Interferon. Altevir® possui efeitos antivirais, imunomoduladores, antiproliferativos e antitumorais.
O interferon alfa-2b, interagindo com receptores específicos na superfície celular, inicia uma cadeia complexa de mudanças dentro da célula, incluindo a indução da síntese de uma série de citocinas e enzimas específicas, e interrompe a síntese de RNA viral e proteínas virais em a célula. O resultado dessas alterações é uma atividade antiviral e antiproliferativa inespecífica associada à prevenção da replicação viral na célula, à inibição da proliferação celular e ao efeito imunomodulador do interferon. O interferon alfa-2b estimula o processo de apresentação de antígenos às células imunocompetentes, tem a capacidade de estimular a atividade fagocítica dos macrófagos, bem como a atividade citotóxica das células T e das células “natural killer” envolvidas na imunidade antiviral.
Previne a proliferação celular, especialmente células tumorais. Tem efeito inibitório na síntese de alguns oncogenes, levando à inibição do crescimento tumoral.
Com a administração parenteral do medicamento, são possíveis calafrios, febre, fadiga, dor de cabeça, mal-estar e síndrome semelhante à gripe. Esses efeitos colaterais são parcialmente aliviados com paracetamol ou indometacina.A invenção refere-se à engenharia genética, biotecnologia, medicina, farmacologia. Um novo DNA plasmídico multicópia recombinante pSX50, que codifica a síntese do interferon alfa-2b de leucócitos humanos, cuja expressão está sob o controle de promotores de lactose e triptofano e um terminador de transcrição. Como resultado da transformação de células da cepa receptora E. coli BL21 com DNA plasmídico recombinante pSX50, foi obtida a cepa E. coli SX50 - produtora de interferon alfa-2b humano leucocitário recombinante com produtividade de até 0,9-1,0 g de interferon alfa-2b de 1 litro de meio de cultura. O método de produção de interferon alfa-2b recombinante é baseado no uso de uma cepa recombinante criada de E. coli SX50 e envolve seu cultivo profundo em meio nutriente com teor reduzido de triptofano com adição contínua de substratos nutrientes no processo de biossíntese , destruição mecânica de células de microrganismos em alta pressão, dissolução da proteína agregada em uma solução concentrada de cloridrato de guanidina, seguida de renaturação do interferon em soluções tampão fisiológicas na presença de agentes caotrópicos e sua purificação usando purificação cromatográfica de interferon em três estágios em Resinas quelantes Sepharose Fast Flow imobilizadas com íons Cu +2, cromatografia de troca iônica em resinas de troca iônica como Sephsrose Fast Flow SM e cromatografia de filtração em gel em resinas do tipo Superdex 75. O método permite obter uma substância interferon de mais de 99% pureza de acordo com eletroforese em condições redutoras e não redutoras ao colorir géis com prata e mais de 98% de acordo com RF HPLC e livre de pirogênios (teste LAL) em quantidades de pelo menos 400-800 mg por 1 litro de meio de cultura. 3 n. e 3 cargos salariais, 6 doentes.



A invenção refere-se a medicamentos geneticamente modificados e obtidos biotecnologicamente, nomeadamente a métodos de produção industrial de interferão alfa-2b leucocitário humano recombinante para fins médicos (doravante designado por interferão), bem como a estirpes produtoras de Escherichia coli (E. coli) e DNA plasmídico, que codifica a síntese de interferon.
Os interferons são moléculas de proteína com peso molecular de 15.000 a 21.000 daltons produzidas e secretadas pelas células em resposta a infecções virais ou outros patógenos. Existem três grupos principais de interferons: alfa, beta e gama. Estes grupos em si não são homogéneos e podem conter várias espécies moleculares diferentes de interferão. Assim, foram identificadas mais de 14 variedades genéticas de interferon alfa, que são de interesse e amplamente utilizadas na medicina como agentes antivirais, antiproliferativos e imunomoduladores.
Existem métodos conhecidos para a obtenção de interferão leucocitário humano a partir de leucócitos de sangue de dadores humanos induzidos por vírus e outros indutores (SU1713591, RU 2066188, RU 2080873).
A principal desvantagem destes métodos de produção de interferons é a probabilidade de contaminação do produto final com vírus humanos, como vírus da hepatite B e C, vírus da imunodeficiência, etc.
Atualmente, o método de produção de interferon por síntese microbiológica tem sido reconhecido como mais promissor, o que permite obter o produto alvo com rendimento significativamente maior a partir de materiais de partida relativamente baratos. As abordagens aqui utilizadas permitem criar variantes de um gene estrutural que são ideais para a expressão bacteriana, bem como elementos reguladores que controlam a sua expressão.
Vários desenhos de cepas de Pichia pastoris, Pseudomonas putida e Escherichia coli são usados como microrganismos fonte.
A desvantagem de usar P. pastoris como produtor de interferon (J.N. Garcia, J.A. Aguiar et. al. //High level expression of human IFN-2b in Pichia pastoris.//Biotecnologia Aplicada, 12(3),152-155, 1995 ), As condições de fermentação deste tipo de levedura são extremamente difíceis, sendo necessária a manutenção rigorosa da concentração do indutor, em particular o metanol, durante o processo de biossíntese. A desvantagem de usar Ps. putida (SU1364343, SU1640996, SU1591484, RU1616143, RU2142508) é a complexidade do processo de fermentação em um baixo nível de expressão (10 mg de interferon por 1 litro de meio de cultura). Mais produtivo é o uso de cepas de Escherichia coli (Semin. Oncol., 1997, Iun; 24 (3 Suppl. 9): S9-41-S9-51).
É conhecido um grande número de plasmídeos e cepas de E. coli criados com base neles que expressam interferon: cepas de E. coli ATCC 31633 e 31644 com plasmídeos Z-pBR322 (Psti) HclF-11-206 ou Z-pBR 322(Pstl)/ HclN SN 35 -AHL6 (SU 1764515), cepa de E. coli pINF-AP2 (SU 1312961), cepa de E. coli pINF-F-Pa (AU 1312962), cepa de E. coli SG 20050 com plasmídeo p280/21FN (Kravchenko V.V. e outros. Bioorganic chemistry, 1987, v. 13, no. 9, pp. 1186-1193), cepa E. Coli SG 20050 com plasmídeo pINF14 (SU 1703691), cepa E. coli SG 20050 com plasmídeo pINF16 (RU 2054041) e etc. A desvantagem das tecnologias baseadas no uso dessas cepas é a sua instabilidade, bem como o nível insuficiente de expressão do interferon.
Juntamente com as características das cepas utilizadas, a eficiência do processo depende em grande parte da tecnologia utilizada para o isolamento e purificação do interferon.
Existe um método conhecido para a produção de interferon, que inclui a cultura de células Ps. putida, destruição de biomassa, tratamento com polietilenimina, fracionamento com sulfato de amônio, cromatografia hidrofóbica em fenilsilocromo C-80, fracionamento de pH do lisado, sua concentração e diafiltração, cromatografia de troca iônica em celulose DE-52, eluição em gradiente de pH, íon cromatografia de troca do eluente resultante em celulose SM -52, concentração por passagem através de um cassete de filtro e filtração em gel em Sephadex G-100 (SU 1640996). A desvantagem deste método, além da complexa fermentação em várias etapas, é o processo em várias etapas na obtenção do produto final.
Também existe um método conhecido para a produção de interferon, que inclui o cultivo da cepa de E. coli SG 20050/pIF16 em caldo LB em frascos em agitador termostatado, centrifugação da biomassa, lavagem com solução tampão e tratamento ultrassônico para destruição das células. O lisado resultante é centrifugado, lavado com solução de ureia 3M em tampão, dissolvido em solução de cloreto de guanidina em tampão, tratado com ultrassom, centrifugado, sulfitólise oxidativa, diálise contra ureia 8 M, renaturação e cromatografia final em dois estágios em CM- 52 celulose e Sephadex G-50 (RU 2054041). As desvantagens deste método são a produtividade relativamente baixa das principais etapas do processo de isolamento e purificação. Isto se aplica especialmente ao tratamento ultrassônico do produto, diálise e sulfitólise oxidativa, o que leva à instabilidade no rendimento do interferon, bem como à impossibilidade de utilização deste método para a produção industrial de interferon.
Como análogo mais próximo (protótipo), pode ser indicado um método para obtenção de interferon leucocitário humano, que consiste em cultivar uma cepa recombinante de E. coli, congelar a biomassa resultante a uma temperatura não superior a -70°C, descongelar, destruir células de microrganismos com lisozima, remoção de DNA e RNA pela introdução no lisado de DNAse e purificação da forma insolúvel isolada de interferon por lavagem com uma solução tampão com detergentes, dissolução do precipitado de interferon em uma solução de cloridrato de guanidina, renaturação e purificação de íons em uma etapa cromatografia de troca. A estirpe SS5 de E. coli obtida utilizando o plasmídeo recombinante pSS5 contendo três promotores: Plac, Pt7 e P trp, e o gene do interferão alfa com substituições de nucleótidos introduzidas é utilizada como produtor.
A expressão do interferão pela estirpe SS5 de E. coli contendo este plasmídeo é controlada por três promotores: Plac, Pt7 e Ptrp. O nível de expressão de interferon é de cerca de 800 mg por 1 litro de suspensão celular (RU 2165455).
A desvantagem desse método é a baixa eficiência tecnológica do uso da destruição enzimática de células, DNA e RNA do microrganismo e purificação cromatográfica de interferon em uma etapa. Isto causa instabilidade no processo de liberação do interferon, leva à diminuição da sua qualidade e limita a possibilidade de utilização do esquema acima para a produção industrial de interferon. As desvantagens deste plasmídeo e da cepa baseada nele são o uso no plasmídeo de um forte promotor não regulamentado do fago T7 na cepa de E. coli BL21 (DE3), na qual o gene da RNA polimerase T7 está localizado sob o promotor de o operon lac e que está sempre “fluindo”. Consequentemente, a síntese do interferon ocorre continuamente na célula, o que leva à dissociação do plasmídeo e à diminuição da viabilidade das células da cepa e, como resultado, à diminuição do rendimento do interferon.
O objetivo desta invenção é construir uma cepa produtora industrial recombinante de E. coli usando um novo DNA plasmidial recombinante com alto nível de biossíntese de interferon, e desenvolver uma tecnologia industrial eficaz para produzir uma substância interferon para uso médico, correspondente em qualidade à "Farmacopeia Europeia" para a substância interferão alfa-2b.
Este problema foi resolvido com a criação do DNA do plasmídeo recombinante pSX50 e da cepa de Escherichia coli SX50, depositada na Coleção Russa de Cepas Industriais do Instituto de Pesquisa Estadual de Genética da Empresa Unitária do Estado Federal, número VKPM B-8550,
bem como um método para produção de interferon alfa-2b recombinante, baseado no uso de uma cepa recombinante de E. coli SX50 e envolvendo seu cultivo profundo em meio nutriente com teor reduzido de triptofano com adição contínua de substratos nutrientes no processo de biossíntese, destruição mecânica de células de microrganismos em alta pressão, dissolução de proteína agregada em uma solução concentrada de cloridrato de guanidina, seguida de renaturação de interferon em soluções tampão fisiológicas na presença de agentes caotrópicos e purificação cromatográfica de interferon em três estágios em resinas como como Chelating Sepharose Fast Flow, imobilizado com íons Cu +2, cromatografia de troca iônica em resinas de troca iônica como CM Sepharose Fast Flow e cromatografia de filtração em gel em resinas como Superdex 75.
De acordo com a invenção, é proposto um novo DNA plasmídico multicópia recombinante pSX50, que codifica a síntese do interferon alfa-2b de leucócitos humanos, cuja expressão está sob o controle dos promotores de lactose e triptofano e de um terminador de transcrição. O plasmídeo pSX50 possui 3218 pares de bases (pb) e é caracterizado pela presença dos seguintes fragmentos:
A sequência do nucleotídeo 1 ao nucleotídeo (nt) 176 inclui um fragmento de DNA de 176 pb contendo o promotor de triptofano (P trp);
Sequência de 177 nt. a 194 n. inclui um fragmento de DNA sintético de 18 pb contendo a sequência Shine Delgarno, responsável pelo início da tradução;
Sequência de 195 nt. a 695 n. inclui um fragmento de DNA de 501 pb de tamanho contendo a sequência do gene do interferon com as seguintes substituições de nucleotídeos: na posição 37, substituição de A por C, na posição 39, substituição de G por T, na posição 40, substituição de A por C, na posição 42, substituição de G por T, na posição 67, substituição de A por C, na posição 69, substituição de G por T, na posição 70, substituição de A por C, na posição 72, substituição de A por T, na posição 96, substituição de G por A, na posição 100, substituição de A por C, na posição 102, substituição de A por T, na posição 114, substituição de A por C, na posição 120, substituição de C com G, na posição 126, substituição de G por A, na posição 129, substituição de G por A, na posição 330, substituição de C por G, na posição 339 substituindo G por A, na posição 342 substituindo G por A, em posição 487 substituindo A por C, na posição 489 substituindo A por T, na posição 495 substituindo G por A;
Sequência de 696 nt. de acordo com 713 n. inclui um fragmento de DNA sintético de 18 pb contendo um poliligante sintético;
Sequência de 714 nt. a 1138 n. inclui um fragmento de DNA do plasmídeo pKK223-3 com 4129 nt. a 4553 n. 425 pb de tamanho, contendo a sequência do terminador de transcrição estrito rrnBT 1 T 2;
Sequência de 1139 b. a 1229 n. inclui um fragmento de DNA do plasmídeo pUC19 com 2.487 nt. a 2577 n. 91 pb de tamanho, contendo o promotor do gene da β-lactomase (gene de resistência à ampicilina - Amp R);
Sequência de 1230 a.C. até 2045 n. inclui um fragmento de DNA do plasmídeo pUC4K com 720 nt. até 1535 DC 816 pb de tamanho, contendo a região estrutural do gene kan;
Sequência de 2046 b. a 3218 n. inclui um fragmento de DNA do plasmídeo pUC19 de 1625 a 453 nt. Tamanho de 1173 pb, contendo a sequência responsável pela replicação do plasmídeo (ori) e promotor lac (P lac).
As Figuras 1-5 mostram diagramas de construção e um mapa físico do plasmídeo pSX50.
A Figura 6 mostra a sequência nucleotídica completa determinada para o plasmídeo pSX50.
A cepa SX50 de Escherichia coli foi obtida pela transformação de células BL21 de Escherichia coli com o plasmídeo pSX50 usando tecnologia tradicional de engenharia genética. A cepa E.Coli SX50 é caracterizada pelas seguintes características.
Características culturais e morfológicas
As células são pequenas, retas, espessadas em forma de bastonete, gram-negativas e sem esporos. As células crescem bem em meios nutrientes simples. Ao crescer em ágar Difco, formam-se colônias redondas, lisas, convexas, turvas, brilhantes e cinzentas com bordas lisas. Quando cultivado em meio líquido (em meio mínimo com glicose ou em caldo LB), forma-se uma turbidez intensa e uniforme.
Características físicas e biológicas
Aeróbio. A faixa de temperatura para crescimento é de 4-42°C com um pH ideal de 6,5-7,5.
Tanto os sais minerais na forma de amônio e nitrato, quanto os compostos orgânicos na forma de aminoácidos, peptona, triptona, extrato de levedura, etc. são utilizados como fonte de nitrogênio.
Aminoácidos, glicerol e carboidratos são usados como fonte de carbono. Resistência a antibióticos. As células apresentam resistência à canamicina (até 100 μg/ml).
A cepa 8X50 de Escherichia coli é produtora de interferon.
Método, condições e composição do meio de armazenamento de cepas
Em L-arape com adição de canamicina na concentração de 20 mcg/ml sob óleo, em caldo L contendo 15% de glicerol e antibióticos apropriados em ampolas a uma temperatura de -70°C, no estado liofilizado em ampolas a uma temperatura de mais 4°C.
A cepa SX50 de Escherichia coli foi identificada de acordo com Bergey's Guide (1974) como uma cepa da espécie Escherichia coli.
Método para produção industrial de interferon alfa-2b
Uma característica do método proposto é o desenvolvimento de uma tecnologia que permite isolar o interferon de uma forma insolúvel que se acumula durante a fermentação, o que permite simplificar significativamente o esquema tecnológico do processo de isolamento e aumentar o rendimento do produto alvo.
O método consiste no cultivo da cepa Escherichia coli SH50 em meio nutriente, com adição constante de substratos nutrientes, preferencialmente glicose e extrato de levedura, no processo de biossíntese, preferencialmente com teor reduzido de triptofano, destruição mecânica de células de microrganismos em alta pressão 700-900 bar, dissolução do interferon em solução tampão de cloridrato de guanidina, renaturação do interferon em soluções tampão fisiológicas na presença de agentes caotrópicos, seguida de purificação cromatográfica em três estágios do interferon em resinas quelantes do tipo Sepharose Fast Flow imobilizadas com Cu +2 íons, cromatografia de troca iônica em resinas de troca iônica do tipo CM Sepharose Fast Flow e cromatografia de filtração em gel em resinas como Superdex 75.
As condições ideais para a realização de etapas individuais de produção de interferon são as seguintes:
A fermentação é realizada com adição contínua de substratos ao longo do processo, o que provoca alto nível de expressão de interferon;
A destruição celular é realizada num desintegrador do tipo Gaulin a uma pressão de 900 bar;
A remoção dos componentes celulares solúveis (DNA, RNA, proteínas, lipopolissacarídeos, etc.) é realizada lavando a forma insolúvel do interferon com soluções tampão contendo detergentes (Triton XI00, uréia, etc.);
O precipitado resultante contendo interferão é dissolvido numa solução tampão de cloridrato de guanidina 6 M;
A renaturação do interferon é realizada em solução tampão fisiológica contendo agentes caotrópicos;
A purificação cromatográfica do interferon em três estágios é realizada em Chelating Sepharose Fast Flow, imobilizado com íons Cu +2, em resina de troca catiônica SM Sepharose Fast Flow e cromatografia de filtração em gel em resina do tipo Superdex 75;
Após cada purificação cromatográfica, a filtração esterilizante é realizada através de filtros isentos de pirogênio com poros de 0,22 mícrons.
O rendimento de interferon como resultado do uso do método descrito é de aproximadamente 400-800 mg de interferon por 1 litro de meio de cultura. A qualidade do produto resultante cumpre as normas e requisitos da "Farmacopeia Europeia" para a substância interferão alfa-2b.
Diferenças significativas entre o método proposto e o protótipo são:
A utilização de um desenho de cepa com maior produtividade, que permite obter maior quantidade de interferon a partir de 1 litro de meio de cultura durante a biossíntese;
A utilização de destruição mecânica eficaz da biomassa celular, que permite obter um extrato mais puro da forma insolúvel do interferon em menor tempo e com menores perdas;
A utilização de soluções tampão fisiológicas durante a renaturação na presença de agentes caotrópicos permite aumentar o rendimento da forma corretamente renaturada de interferon;
A purificação cromatográfica de interferon em três estágios permite obter uma substância interferon com pureza superior a 99% de acordo com eletroforese em condições redutoras e não redutoras na coloração de géis com prata e mais de 98% de acordo com RF HPLC e praticamente isenta de pirogênios (teste LAL).
A essência e as vantagens do grupo reivindicado de invenções são ilustradas pelos exemplos a seguir.
Exemplo 1. Construção do plasmídeo recombinante pSH50
O método para construir o plasmídeo pSX50 inclui as seguintes etapas:
Construção do plasmídeo vetorial pSX10;
1. construção do plasmídeo pSX3 (2641 pb)
2. construção do plasmídeo vetorial pSX10 (2553 pb)
Construção do plasmídeo recombinante pSX41 (3218 pb);
Construção do plasmídeo recombinante pSX43 (3218 pb);
Construção do plasmídeo recombinante pSX45 (3218 pb);
Construção do plasmídeo recombinante pSX50 (3218 pb).
Construção do plasmídeo vetorial pSX10
O plasmídeo vetorial pSX10 é um vetor pUC19 no qual a sequência codificadora do gene da beta lactomase, que fornece resistência à ampicilina, é substituída pela sequência codificadora do gene kan e contém o terminador de transcrição do plasmídeo pKK223-3.
A construção do plasmídeo vetorial pSS10 é realizada em duas etapas:
Preparação do plasmídeo pSX3 (2641 pb), que é o plasmídeo pUC19, no qual a região codificadora do gene amp é substituída pela região codificadora do gene kan;
Preparação do plasmídeo vetorial pSX10 (2553 pb), que é um plasmídeo pSX3 no qual um fragmento de DNA que codifica o terminador de transcrição rBT 1 T 2 é inserido atrás do sítio BamHI.
Para obter o plasmídeo pSX3, são realizadas cinco rodadas de amplificação de DNA usando PCR (reação em cadeia da polimerase). Durante a primeira ronda, utilizando ADN do plasmídeo pUC19 como modelo, é amplificado um fragmento de ADN de 1828 pb de tamanho. (fragmento PU1-PU2) usando primers:
Esta e as reações de PCR subsequentes são realizadas sob as seguintes condições: Tis-HCl 20 mM, pH 8,8, (NH 4) 2 SO 4 10 mM, KCl 10 mM, MgCl 2 2 tM, Triton X100 a 0,1%, 0,1 mg/ml BSA, 0,2 mM de cada dNTP, 1,25 unidades. Pfu DNA polimerase, 100 ng de DNA. O processo de amplificação consiste nas seguintes etapas: aquecimento a 95°C por 5 min, 35 ciclos de PCR (30 segundos a 95°C, 30 segundos a 56°C, 2 minutos a 72°C) e incubação por 10 minutos a 72°C. Após amplificação (e após amplificações subsequentes), o fragmento de DNA é purificado por eletroforese em gel de agarose a 1%. Durante a segunda e terceira rondas, utilizando ADN do plasmídeo pUC4K como modelo, é amplificado um fragmento de ADN de 555 pb. (fragmento KM1-KM2) usando primers:
e amplificação de um fragmento de DNA de 258 pb. (KMZ-KM4) de primers
Na quinta rodada de PCR, os fragmentos (PU1-PU2) e (KM1-KM4) são combinados sob as seguintes condições: aquecimento a 95°C por 5 min, 5 ciclos de PCR (30 seg 95°C, 30 seg 56°C , 10 min 72°C) e incubação por 10 min a 72°C. O DNA obtido após a última PCR é diretamente transformado em células de E. coli cepa DH5 e plaqueado em meio LA contendo 20 μg/ml de canamicina. Após incubação durante 12 horas a 37°C, os clones são eliminados, o ADN plasmídico é isolado e é realizada a análise de restrição. Como resultado, é obtido o plasmídeo pSX3 com tamanho de 2.641 pb.
Para obter o plasmídeo vetorial pSX10, são realizadas três rodadas de amplificação de DNA usando PCR. Na primeira rodada, usando o DNA do plasmídeo pSX3 como modelo, um fragmento de DNA de 2.025 pb é amplificado. (fragmento 10.1-10.2) usando primers:
Durante a segunda ronda, utilizando o ADN do plasmídeo pKK223-3 como molde, é amplificado um fragmento de ADN de 528 pb de tamanho. (fragmento KK1-KK2) usando primers:
Na terceira rodada de PCR, os fragmentos (10,1-10,2) e (KK1-KK2) são combinados sob as seguintes condições: aquecimento a 95°C por 5 minutos, 5 ciclos de PCR (30 segundos a 95°C, 30 segundos a 56°C , 10 min 72°C) e incubação por 10 min a 72°C. O DNA obtido após a última PCR é diretamente transformado em células de E. coli cepa DH5 e plaqueado em meio LA contendo 20 μg/ml de canamicina. Após incubação durante 12 horas a 37°C, os clones são eliminados, o ADN plasmídico é isolado e é realizada a análise de restrição. Como resultado, obtém-se o plasmídeo pSX10 com um tamanho de 2553 pb.
Construção do plasmídeo recombinante pSX41
O plasmídeo recombinante pSX41 é um fragmento de DNA Hind III - BamHI do plasmídeo vetorial pSX3 (2529 pb), fragmento de DNA Hind III - EcoRI de 168 pb, codificando o promotor do operon triptofano de E. coli (P trp), EcoRI-XbaI um sintético Fragmento de DNA de 20 pb que codifica a sequência SD (Shine-Delgarno) e um fragmento de DNA XbaI-BamHI de 501 pb que codifica o gene do interferon alfa 2b humano.
Para obter o fragmento de DNA Hind III - BamHI do plasmídeo vetorial pSX3 (2529 pb), o DNA do plasmídeo pSX3 é tratado com as enzimas de restrição HindIII e BamHI, seguido de purificação eletroforética em gel de agarose a 1%. O fragmento de DNA Hind III EcoRI de 168 pb que codifica o promotor do operon triptofano (P trp) é obtido por PCR usando DNA total de E. coli como modelo e primers TRP1 e PRP2, seguido de tratamento do fragmento amplificado com restrição Hindl e EcoRI enzimas:
Para obter EcoRI-Xbal um fragmento de DNA sintético de 20 pb que codifica a sequência SD (Shine-Delgarno), são sintetizados os seguintes oligonucleotídeos complementares:
O fragmento de DNA XbaI-BamIII de 501 pb de tamanho, que codifica o gene do interferon alfa 2b humano, é obtido por PCR usando DNA humano total como modelo e iniciadores IFN1 e IFN2, seguido pelo processamento do fragmento amplificado com enzimas de restrição Xbal e BamIII:
Em seguida, os fragmentos purificados eletroforeticamente são combinados, ligados com a enzima ligase do fago T4, o DNA é transformado em células da cepa E. coli DH5 e plaqueado em meio LA contendo 20 μg/ml de canamicina. Após incubação durante 12 horas a 37°C, os clones são eliminados, o ADN plasmídico é isolado, a análise de restrição é realizada e a estrutura primária do ADN é determinada. Como resultado, obtém-se o plasmídeo pSX41 com um tamanho de 3218 pb. Em seguida, é realizada mutagénese passo a passo do gene do interferão para aumentar o nível de expressão do produto alvo. A mutagênese do gene do interferon consiste na substituição de tripletos raramente encontrados em E. coli, codificando os aminoácidos correspondentes, por tripletos frequentemente encontrados em E. coli, codificando os mesmos aminoácidos. A mutagênese do DNA do gene do interferon é realizada pelo método PCR.
Construção do plasmídeo recombinante pSX43
Para obter o plasmídeo recombinante pSX43, uma rodada de amplificação do DNA é realizada por PCR usando o DNA do plasmídeo pSX41 como molde e os iniciadores IFN3 e IFN4:
A PCR é realizada sob as seguintes condições: aquecimento a 95°C durante 5 min, 20 ciclos de PCR (30 segundos a 95°C, 30 segundos a 56°C, 10 minutos a 72°C) e incubação durante 20 minutos a 72°C. O DNA obtido após PCR é diretamente transformado em células da cepa DH5 de E. coli e plaqueado em meio LA contendo 20 μg/ml de canamicina. Após incubação durante 12 horas a 37°C, os clones são eliminados, o ADN plasmídico é isolado, a análise de restrição é realizada e a estrutura primária do ADN é determinada. Como resultado, obtém-se o plasmídeo pSX43 com um tamanho de 3218 pb.
Construção do plasmídeo recombinante pSX45
Para obter o plasmídeo recombinante pSX45, uma rodada de amplificação do DNA é realizada por PCR usando o DNA do plasmídeo pSX43 como molde e os iniciadores IFN5 e IFN6:
A PCR é realizada sob as seguintes condições: aquecimento a 95°C durante 5 min, 20 ciclos de PCR (30 segundos a 95°C, 30 segundos a 56°C, 10 minutos a 72°C) e incubação durante 20 minutos a 72°C. O DNA obtido após PCR é diretamente transformado em células da cepa DH5 de E. coli e plaqueado em meio LA contendo 20 μg/ml de canamicina. Após incubação durante 12 horas a 37°C, os clones são eliminados, o ADN plasmídico é isolado, a análise de restrição é realizada e a estrutura primária do ADN é determinada. Como resultado, obtém-se o plasmídeo pSX45 com um tamanho de 3218 pb.
Construção do plasmídeo recombinante pSX50.
Para obter o plasmídeo recombinante pSX50, uma rodada de amplificação do DNA é realizada por PCR usando o DNA do plasmídeo pSX45 como molde e os iniciadores IFN7 e IFN8:
A PCR é realizada sob as seguintes condições: aquecimento a 95°C durante 5 min, 20 ciclos de PCR (30 segundos a 95°C, 30 segundos a 56°C, 10 minutos a 72°C) e incubação durante 20 minutos a 72°C. O DNA obtido após PCR é diretamente transformado em células da cepa DH5 de E. coli e plaqueado em meio LA contendo 20 μg/ml de canamicina. Após incubação durante 12 horas a 37°C, os clones são eliminados, o ADN plasmídico é isolado, a análise de restrição é realizada e a estrutura primária do ADN é determinada. Como resultado, obtém-se o plasmídeo pSX50 com um tamanho de 3218 pb.
Exemplo 2. Preparação da cepa E. coli SX50 - produtora de interferon
A cepa E. coli SX50 produtora de interferon é obtida pela transformação de células da cepa E. coli BL21 com o plasmídeo recombinante pSX50. A estirpe produtora de interferão é cultivada num fermentador de 30 litros até uma densidade óptica de 25,0-30,0 p.u. em meio M9 contendo 1% de hidrolisado ácido de caseína (Difco), 1% de glicose, 40 µg/ml de canamicina, a uma temperatura de 38-39°C. Durante o processo de fermentação, é realizada uma adição contínua de substrato nutriente por meio de um controlador gravimétrico.
Exemplo 3. Método para isolar interferon da cepa SX50 de E. coli
O interferon foi obtido em 4 etapas:
Estágio 1. Cultivo de E. coli cepa SX50.
Etapa 2. Isolamento e purificação da forma insolúvel de interferon.
Etapa 3. Dissolução e renaturação do interferon.
Etapa 4. Purificação cromatográfica de interferon.
Estágio 1. Cultivo da cepa E. coli SX50
Inóculo cultivado de E. coli cepa SX50 em um volume de 3 litros de meio LB rico por 12 horas a 26°C é introduzido assepticamente em um fermentador contendo 27 litros de meio estéril contendo M9, 1% de hidrolisado ácido de caseína, 1% de glicose, MgCl2 1 mM, CaCl2 0,1 mM, canamicina 40 mg/ml. O cultivo em fermentador é realizado a uma temperatura de 38-39°C, mantendo um pH de 7±0,15 por titulação automática com solução de hidróxido de sódio a 40%. A concentração de oxigênio dissolvido na faixa de (50±10)% de saturação é mantida alterando a velocidade do agitador de 100 para 800 rpm e o suprimento de ar de 1 para 15 l/min. A concentração de substratos, em particular glicose e extrato de levedura, é medida durante a fermentação e sua concentração é mantida variando a taxa de fornecimento de soluções concentradas através de bombas peristálticas usando um controlador gravimétrico.
A acumulação de interferão na forma insolúvel é monitorizada utilizando microscopia de contraste de fase, electroforese em gel de poliacrilamida a 15% (SDS-PAAG) e cromatografia de alto desempenho de fase reversa (RF HPLC). A fermentação é interrompida quando a densidade óptica máxima (~ 25-30 p.u.) é atingida e a síntese de interferon é interrompida. Ao final da fermentação, o líquido cultural é separado por centrifugação em rotor de fluxo a uma velocidade de rotação de 5.000-10.000 rpm. A biomassa é embalada em sacos plásticos e congelada a 70°C negativos.
Etapa 2. Isolamento e purificação da forma insolúvel de interferon
300-400 g de biomassa congelada de E. coli estirpe SX50 são suspensos em 3000 ml de tampão 1 (Tris-HCl 20 mM, pH 8,0, EDTA 10 mM, Triton X100 a 0,1%). A suspensão é passada através de um homogeneizador de fluxo tipo Gaulin, mantida a uma pressão de 900 bar e centrifugada em um rotor de fluxo a 15.000 rpm. O precipitado resultante é lavado sob condições semelhantes sequencialmente com tampões 2 (Tris-HCl 20 mM, pH 8,0, EDTA 1 mM, ureia 3 M) e tampão 3 (Tris-HCl 20 mM pH 8,0, EDTA 1 mM) e finalmente o interferon o precipitado é suspenso em 200 ml de tampão 3. Neste caso, o tempo para isolamento e purificação da forma insolúvel do interferon não é superior a 5 horas.
Etapa 3. Dissolução e renaturação do interferon
À suspensão da forma insolúvel de interferon obtida na etapa anterior, adicionar cloridrato de guanidina seco a uma concentração de 6 M, adicionar ditiotreitol a uma concentração de 50 mM, Tris-HCl pH 8,0 a uma concentração de 50 mM, NaCl a um concentração de 150 mM e Triton X100 a uma concentração de 0,1%, incubar à temperatura ambiente durante 2 horas. O material não dissolvido é separado por filtração esterilizante através de membranas com diâmetros de poros de 0,22 mícrons.
A renaturação do interferão é realizada diluindo lentamente a solução resultante 100-200 vezes com tampão 4 (Tris-HCl 20 mM pH 8,0, NaCl 100 mM, EDTA 0,1 mM). Após o que a mistura de renaturação é incubada com agitação constante durante 12-15 horas a uma temperatura de 4-8°C. O sulfato de magnésio é então adicionado a uma concentração de 1 mM e o material agregado é removido por filtração esterilizante através de um filtro de membrana com um diâmetro de poro de 0,22 mícron.
Etapa 4. Purificação cromatográfica de interferon
A purificação cromatográfica do interferon é realizada em três etapas.
1. O interferão renaturado resultante é primeiro purificado utilizando cromatografia de afinidade numa resina Chelating Sepharose Fast Flow (Amersham Biosciences) imobilizada com iões Cu+2. Para fazer isso, a solução de interferon é aplicada a uma coluna com Cu+2 Chelating Sepharose Fast Flow e o interferon é eluído com um tampão de ácido cítrico 0,1 M, pH 2,2.
2. No segundo estágio de purificação cromatográfica, a solução de interferon é aplicada a uma resina de troca catiônica do tipo CM Sepharose Fast Flow (Amersham Biosciences) e o interferon é eluído com um gradiente de soluções (0,0-0,5 M NaCl) em 50 mM. Tampão Na(CH 3 COO), pH 5,5.
3. A purificação da forma monomérica de interferon a partir dos restos de formas poliméricas de interferon é realizada no terceiro estágio de purificação de interferon por filtração em gel em resina Superdex 75 (Amersham Biosciences). A cromatografia é realizada num tampão de Na(CH3COO) 50 mM, pH 5,0, contendo NaCl 0,15 M.
O método descrito para isolar e purificar o interferon permite obter 4-8 g de interferon altamente purificado em um ciclo de isolamento durante 7-10 dias a partir de biomassa obtida de 10 litros de meio de cultura. A qualidade do interferão resultante cumpre integralmente os requisitos da “Farmacopeia Europeia” para a substância interferão alfa-2b, nomeadamente:
A concentração de interferon não é inferior a 2×10 8 UI/ml;
A atividade específica do interferon não é inferior a 2,0×10 8 UI/mg;
A pureza eletroforética do fármaco é de pelo menos 99% sob condições redutoras e não redutoras ao colorir géis com prata;
O ponto isoelétrico do interferon isolado está na região de pH 5,8-6,3;
O mapa peptídico do interferon isolado não é fundamentalmente diferente do mapa peptídico do interferon alfa 2b CRS padrão europeu;
Como se segue dos exemplos dados, o grupo de invenções reivindicado permite obter interferon alfa-2b com alto rendimento utilizando uma tecnologia relativamente simples e confiável.
1. DNA plasmidial recombinante pSX50, que codifica a síntese do interferon alfa-2b humano recombinante, caracterizado por possuir tamanho de 3218 pares de bases (pb) e ser composto pelos seguintes fragmentos: a sequência de 1 a 176 nucleotídeos (pb) inclui um fragmento de DNA de 176 pb contendo o promotor de triptofano (P trp), sequência de 177 a 194 nt. inclui um fragmento de DNA sintético de 18 pb de tamanho contendo a sequência Shine Delgarno, responsável pelo início da tradução, sequência de 195 a 695 nt. inclui um fragmento de DNA de 501 pb contendo o gene do interferon alfa-2b com substituições de nucleotídeos: 37 (A>C), 39 (G>T), 40 (A>C), 42 (G>T), 67 (A> C), 69 (G>T), 70 (A>C), 72 (A>T), 96 (G>A), 100 (A>C), 102 (A>T), 114 (A>С ), 120 (C>G), 126 (G>A), 129 (G>A), 330 (C>G), 339 (G>A), 342 (G>A), 487 (A>C) , 489 (A>T), 495 (G>A), sequência de 696 a 713 nt. inclui um fragmento de DNA sintético de 18 pb contendo um poliligante sintético, sequência de 714 a 1138 nt. inclui um fragmento de DNA do plasmídeo pKK223-3 de 4129 a 4553 nt. 425 pb de tamanho, contendo a sequência do terminador de transcrição estrito rrnBT 1 T 2, sequência de 1139 a 1229 nt. inclui um fragmento de DNA do plasmídeo pUC19 de 2.487 a 2.577 nt. Tamanho de 91 pb, contendo o promotor do gene -lactomase (gene de resistência à ampicilina -Amp R), sequência de 1230 a 2045 nt. inclui um fragmento de DNA do plasmídeo pUC4K com 720 nt. até 1535 DC Tamanho de 816 pb, contendo a região estrutural do gene kan, sequência com 2.046 pb. a 3218 n. inclui um fragmento de DNA do plasmídeo pUC19 de 1625 a 453 nt. Tamanho de 1173 pb, contendo a sequência responsável pela replicação do plasmídeo (ori) e promotor lac (P lac).
2. A cepa bacteriana Eschcerichia coli SX50 transformada com o plasmídeo recombinante de acordo com a reivindicação 1 é produtora de interferon alfa-2b leucocitário humano recombinante.
3. Método para produção de interferon alfa-2b humano, incluindo cultivo da cepa Escherichia coli SX5 de acordo com a reivindicação 2 em meio nutriente com adição constante de substratos nutrientes durante o processo de biossíntese, destruição mecânica de células de microrganismos a uma pressão de 700- 900 bar, dissolução de interferon em solução tampão de cloridrato de guanidina, renaturação de interferon em soluções tampão fisiológicas na presença de agentes caotrópicos, purificação cromatográfica de interferon em três estágios em resinas quelantes do tipo Sepharose Fast Flow imobilizadas com íons Cu +2, troca iônica cromatografia em resinas de troca iônica do tipo CM Sepharose Fast Flow e cromatografia de filtração em gel em resinas do tipo Superdex 75.
4. Método de acordo com a reivindicação 3, caracterizado pelo fato de que o cultivo é realizado em meio nutriente com teor reduzido de triptofano com adição contínua de substratos nutrientes, preferencialmente glicose e extrato de levedura.
5. Método de acordo com a reivindicação 3, caracterizado pelo fato de que antes de dissolver o interferon, ele é purificado pela remoção de componentes celulares solúveis, incluindo DNA, RNA, proteínas, lipopolissacarídeos, lavagem com soluções tampão contendo detergentes como Triton XI 00, ureia.
6. Método de acordo com a reivindicação 3, caracterizado pelo fato de que após cada purificação cromatográfica, a filtração esterilizante é realizada através de filtros com tamanhos de poros de 0,22 mícrons.

Em 2018, o Jejum da Natividade começará em 28 de novembro. Durante este período, os crentes ortodoxos se preparam para celebrar o Natal...

Constituir família é o sonho da maioria das mulheres. Eles querem ter um marido amoroso e um monte de filhos. Mas nem sempre é um relacionamento...

Este artigo contém: a oração mais poderosa pelo divórcio - informações retiradas de todos os cantos do mundo, da rede eletrônica e...

Site de informações sobre ícones, orações, tradições ortodoxas. Oração pelos escândalos e brigas na família, com o marido, com os filhos...

O que seria do Ano Novo sem champanhe, tangerinas, Olivier, formol e o “arenque com casaco de pele” preferido de todos. Com o último...

Vamos preparar os ingredientes necessários para os biscoitos. A primeira coisa a fazer é colocar a água para ferver. Nós precisamos...

É possível cadastrar funcionário para o cargo de diretor financeiro - contador-chefe? O contador-chefe afirma...

O chefe de uma pequena empresa pode gerenciar facilmente o orçamento de forma independente. VERIFICADO! Se você conseguir...

A criação de novos projetos envolve uma justificação económica preliminar para a sua viabilidade, posterior...
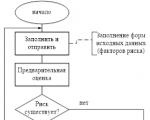
O reporte é gerado pelo RM, é acordado (aprovado) pelo Comité de Risco no âmbito do Conselho de Administração e transmitido a...

Na beira de um prado grande, muito grande, em uma longa folha de grama esmeralda vivia uma pequena joaninha. O pequeno de Deus...

Hoje em dia é bastante comum as pessoas se voltarem para as estrelas. Com a ajuda de um horóscopo uma pessoa pode descobrir...

Negócios ou amigável. Se você estava perseguindo um estranho, isso significa o seu nível de confiança no sexo feminino...

de acordo com o livro dos sonhos de Freud Se você sonhou que estava pescando, isso significa que na vida real dificilmente você consegue desligar...

Constituir família é o sonho da maioria das mulheres. Eles querem ter um marido amoroso e um monte de filhos. Mas nem sempre é...

Este artigo contém: a oração mais poderosa pelo divórcio - informações retiradas de todo o mundo, eletrônicas...