O que é possível e o que não é possível durante o Jejum da Natividade?
Em 2018, o Jejum da Natividade começará em 28 de novembro. Durante este período, os crentes ortodoxos se preparam para celebrar o Natal...
A policitemia é um aumento no número de células sanguíneas por unidade de volume. Existem dois tipos desta doença: policitemia verdadeira (ou primária) e relativa (ou secundária). Esta doença pode ter várias causas: desde doenças do sistema hematopoiético até um aumento fisiológico de glóbulos vermelhos durante a hipóxia.
A policitemia vera é uma doença associada a alterações tumorais benignas no sistema hematopoiético. Observa-se divisão excessiva de células da linhagem mieloide, principalmente no pool de hemácias. Um grande número de glóbulos vermelhos maduros, plaquetas e neutrófilos são liberados simultaneamente no sangue periférico.
A policitemia (CID-10 codifica D 45) leva a alterações na pressão oncótica e aumento da viscosidade do sangue. Por causa disso, a circulação sanguínea nos vasos periféricos fica mais lenta e coágulos sanguíneos e lama podem se formar nos capilares. Em última análise, órgãos e tecidos estão sujeitos à falta de oxigênio e à isquemia.
A primeira menção desta doença apareceu em 1892. Dez anos depois, foi feita uma suposição sobre as causas dessa condição e, ainda mais tarde, ela foi incluída na classificação como uma nosologia separada. Na maioria das vezes, adultos e idosos sofrem de policitemia. Via de regra, os sintomas da doença não apresentam sintomas há muito tempo ou são leves e não preocupam.

As causas da policitemia não são totalmente conhecidas. Os cientistas apresentaram várias teorias sobre isso. O mais confiável deles afirma que as células-tronco sofrem alterações como resultado de mutações. A medicina moderna não sabe por que ocorrem as próprias mutações. Mas o fato de as coordenadas exatas das mudanças terem sido descobertas mostra o avanço que foi feito no estudo desta doença.
Nos estágios iniciais, a doença se manifesta de forma fraca e os pacientes procuram ajuda assim que surgem complicações. Via de regra, trata-se de uma infinidade e de sintomas associados à trombose:
Além de todos os sintomas acima, os pacientes podem queixar-se de deterioração do estado geral, falta de ar, fadiga, tontura, zumbido, dor de cabeça, aumento da pressão arterial e até diminuição da visão.

A policitemia vera é caracterizada não apenas por um complexo de sintomas subjetivos, mas também pelos resultados de estudos objetivos. Em um exame de sangue geral, será observado um número aumentado de glóbulos vermelhos (até 6-8 milhões por mm cúbico com uma norma de 4), juntamente com o número de glóbulos vermelhos, a hemoglobina aumenta, mas o indicador de cor, em pelo contrário, diminui. Porque a hemoglobina acaba dispersa em um grande conjunto de células.
O volume total de sangue é mais que duplicado, o hematócrito muda acentuadamente para os elementos celulares e atinge 65% ou mais. Na forma periférica, começam a aparecer formas jovens de células sanguíneas - reticulócitos. Se normalmente são até um ppm, então na doença de Vaquez esse número oscila entre 15 e 20. Isso indica um aumento patológico na regeneração dos glóbulos vermelhos. Às vezes, células blásticas também podem ser encontradas no esfregaço. Neste caso, o diagnóstico de leucemia pode ser feito erroneamente.
Na fórmula leucocitária, há um desvio acentuado para a esquerda devido às células em banda, bem como um aumento de neutrófilos e eosinófilos. A contagem de plaquetas pode chegar a 600 milhões. Neste contexto, observa-se uma desaceleração da taxa de hemossedimentação para um milímetro por hora.

A policitemia vera se manifesta não apenas e não tanto como sintomas, mas como complicações. Na maioria das vezes, são causadas pelo aumento da formação de trombos nos vasos que irrigam o sistema nervoso central, os órgãos digestivos e as extremidades inferiores. Isto se manifesta por ataques cardíacos e derrames, cirrose hepática, trombose venosa profunda aguda e crônica.
Ao mesmo tempo, você tem que lidar com as consequências da trombose e do sangramento. Devido à isquemia constante, manchas de necrose e úlceras aparecem em áreas delgadas das mucosas e da pele, que começam a sangrar com a irritação mecânica.
Devido ao aumento do ácido úrico, os pacientes geralmente apresentam urolitíase, inflamação da vesícula biliar e esclerose da substância renal.

A policitemia é uma doença polietiológica cujo diagnóstico é bastante difícil. Para garantir que o diagnóstico esteja correto, o médico deve:
Avaliar exame clínico de sangue, parâmetros bioquímicos e hemograma;
Veja os sinais visuais característicos da doença (cor da pele, baço aumentado);
Realize testes para aumento da formação de trombos.
A doença de Vaquez é um diagnóstico de exclusão. Inicialmente, o especialista assume patologias mais comuns e deve certificar-se de que elas não existem. Estes incluem, por exemplo, hipóxia e ingestão descontrolada de vitamina B12. O médico pode precisar de uma punção da medula óssea para confirmar sua conclusão. Isso determinará a função da medula óssea. No total, podem ser necessários cerca de vinte indicadores diferentes para fazer o diagnóstico de policitemia.

Como a policitemia se desenvolve ao longo do tempo? Estágios da patologia, que se diferenciam de acordo com o curso clínico:

Em primeiro lugar, é necessário ter certeza de que o paciente realmente tem policitemia, cujos sintomas e tratamento são discutidos em nossa revisão. Para evitar erros, os médicos desenvolveram todo um algoritmo de ações, a partir do qual é possível fazer um diagnóstico.
Primeiro você precisa coletar uma anamnese e realizar pesquisas. Se o paciente tiver:
Aumento da hemoglobina ou hematócrito;
Baço aumentado;
Trombose das veias hepáticas, então a probabilidade de um diagnóstico correto é alta.
O próximo passo é excluir a policitemia secundária. Para tanto, foram derivados critérios grandes e pequenos.
Critérios grandes:
Critérios menores ou adicionais:
Se o paciente tiver todos os critérios maiores ou dois critérios maiores e dois critérios menores, então com uma probabilidade de 90 por cento pode-se dizer que se trata de policitemia verdadeira.
Para escolher a tática correta, o médico precisa lembrar quais processos fisiopatológicos desencadeiam a policitemia. Os sintomas e o tratamento estão intimamente relacionados. Em primeiro lugar, a terapia visa combater a viscosidade excessiva do sangue e o sangramento.
Para reduzir o número de glóbulos vermelhos, utiliza-se a flebotomia ou, mais simplesmente, a sangria. Apesar de o método ser antigo, senão antigo, ele funciona muito bem neste caso. O excesso de fluido e elementos figurados deixam o leito vascular. Até meio litro de sangue é removido em porções a cada dois a quatro dias até que o hematócrito permaneça dentro de quarenta e cinco por cento e o nível de hemoglobina volte ao normal. Para melhorar o fluxo sanguíneo antes do procedimento, o paciente recebe 400 mililitros de reopoliglucina e cinco mil unidades de heparina.
O segundo método de tratamento é a eritrocitoforese. As tecnologias modernas permitem remover um tipo de célula do corpo, neste caso os glóbulos vermelhos. A quimioterapia também é usada para limitar a produção de células sanguíneas.
Existem várias combinações de métodos selecionados individualmente para cada caso da doença. Isso permite que você alcance o sucesso máximo.
O que espera as pessoas com diagnóstico de policitemia? A previsão não pode ser considerada fatal, mas também não será otimista. No futuro, essas pessoas sofrerão de esclerose da medula óssea e cirrose hepática. Se a doença começar a progredir, tudo terminará em leucemia mieloblástica crônica. Os pacientes vivem em média não mais que dez anos.
Não há diferenças fundamentais em termos patogenéticos entre policitemia verdadeira e falsa (ou relativa). O sangue fica espesso, viscoso, preenche pequenos vasos e leva à trombose. A policitemia secundária é a síndrome de Gaisbeck, também conhecida como pseudopolicitemia de estresse, que pode ser congênita ou adquirida, mas as funções do cérebro vermelho não são alteradas.
Por que aparecem distúrbios hematopoiéticos secundários? Na maioria das vezes, isso está associado a defeitos do sistema cardiovascular que apareceram no útero ou na primeira infância. A presença de doenças hereditárias do sangue em parentes próximos também aumenta a probabilidade de patologia. Mas, via de regra, os pacientes com policitemia secundária não apresentam história familiar. Nesse caso, o médico precisa procurar as causas da falta de oxigênio nos tecidos.
O diagnóstico de policitemia é feito com base na presença de vários sintomas. Infelizmente, não existem patognomônicos entre eles, portanto, diagnosticar esta doença é uma tarefa bastante trabalhosa.
As manifestações mais comuns incluem:
Trombose frequente;
Dor nas articulações e ossos (não associada a lesões);
Dores de cabeça persistentes, tonturas;
Comichão após procedimentos de banho;
Insônia, fadiga, falta de ar;
Perda de peso, sudorese excessiva;
Aumento do fígado do baço.
Esta não é uma lista completa de possíveis queixas dos pacientes. Como pode ser observado na lista, individualmente podem ocorrer no quadro clínico de doenças neurológicas, cardiológicas e reumatológicas.
Assim como na policitemia vera, a hirudoterapia, a doação e os anticoagulantes são utilizados para eliminar os sintomas da doença. A remoção de uma quantidade relativamente pequena de sangue permite reduzir a pressão sistêmica, melhorar a circulação sanguínea nos tecidos e aumentar o fornecimento de oxigênio aos órgãos. Os pacientes sentem tonturas e dores de cabeça, o sono normaliza e a falta de ar desaparece. Mas são medidas paliativas, não podem ser realizadas indefinidamente, por isso o médico ainda deve encontrar a causa da hipóxia e, se possível, eliminá-la o mais rápido possível.

A doença da policitemia não ocorre apenas em adultos. Em casos muito raros, as crianças também podem ser afetadas. Se o feto sofreu hipóxia grave durante a gravidez, após o nascimento terá um número aumentado de glóbulos vermelhos. A falta de oxigênio em uma criança pode ser causada por vários motivos:
A presença de patologia extragenital na mãe (doenças cardíacas, doenças renais, anemia);
Eclampsia;
Síndrome feto-fetal;
- insuficiência placentária e outros.
Após o nascimento, a policitemia pode ser causada por cardiopatias congênitas de difícil correção, como transposição dos grandes vasos ou ducto aórtico patente. As crianças pequenas são mais suscetíveis à hipóxia, por isso o tratamento oportuno desempenha um papel importante. As complicações desta condição incluem hipoplasia da medula óssea e degeneração irreversível dos sistemas nervoso e respiratório.
A policitemia vera é uma doença sanguínea do grupo das leucemias crônicas, caracterizada pela proliferação tumoral (reprodução) principalmente de hemácias. Portanto, esta doença também é chamada de eritremia (das palavras gregas “vermelho” e “sangue”).
A causa da policitemia vera é desconhecida. Supõe-se que nesta doença a regulação do próprio processo de formação de eritrócitos seja inicialmente perturbada.
De acordo com a teoria moderna da hematopoiese, todas as células sanguíneas humanas possuem uma célula precursora. À medida que os descendentes destas células estaminais se dividem e se multiplicam, adquirem características cada vez mais específicas e eventualmente tornam-se glóbulos vermelhos, glóbulos brancos ou plaquetas. Com a eritremia, o equilíbrio do sistema de células sanguíneas muda e começa a formação excessiva e descontrolada de glóbulos vermelhos. Ao mesmo tempo, outras células (leucócitos e plaquetas) também se formam em excesso, mas não tão pronunciadas.
Como resultado, um número aumentado de glóbulos vermelhos aparece no sangue de uma pessoa, não devido a razões externas. Dessa forma, a eritremia difere da eritrocitose, que é a resposta do organismo a um fator externo (por exemplo, falta de oxigênio no ar).
Um aumento no número de glóbulos vermelhos no sangue, bem como uma violação da função plaquetária, leva ao aumento da formação de trombos.
À medida que a doença progride, pode ocorrer a chamada metaplasia mieloide, que se caracteriza pela inibição de todos os germes hematopoiéticos com desenvolvimento.
A aparência de um paciente com policitemia vera é bastante característica. Na maioria das vezes, trata-se de uma pessoa de meia idade ou idosa com excesso de peso. O rosto está vermelho, a esclera está injetada. Os lábios e a língua têm um tom cereja característico. Esses sintomas externos são chamados de “eritrose”.
Os pacientes apresentam sinais de disfunção do sistema nervoso central. Há queixas de zumbido. Desmaios frequentes e... Às vezes, a saúde do paciente piora tanto que ele não consegue realizar nenhum trabalho mental. Preocupa-se com diminuição da memória e atenção, fraqueza, irritabilidade.
Os pacientes geralmente indicam dor no peito. No entanto, essas sensações são mais frequentemente causadas por dor no próprio esterno, como resultado do aumento do suprimento sanguíneo ao tecido. Porém, esses pacientes apresentam alto risco, inclusive de vasos coronários, com o desenvolvimento de e.
As complicações trombóticas podem levar à tromboflebite das veias mesentéricas com o desenvolvimento dos sintomas correspondentes. A ocorrência de acidentes cerebrovasculares também é possível.
Em pacientes com eritremia, é frequentemente diagnosticada, o que está associado a uma violação da regulação nervosa do corpo. Ocasionalmente, desenvolve-se hipertensão arterial (este não é um sintoma muito característico da policitemia).
Junto com a trombose, é frequentemente observada síndrome hemorrágica, associada à tendência a sangrar. Não só os sangramentos nasais são uma preocupação, mas também as hemorróidas, provenientes das veias dilatadas do esôfago, bem como sangramento nas gengivas. Hemorragias subcutâneas também são observadas e facilmente se formam equimoses (hematomas).
Cerca de metade dos pacientes apresentam coceira intensa na pele após tomar banho quente; este é um sintoma característico da eritremia. Alguns pacientes apresentam dor em queimação nas pontas dos dedos, o que também é característico da policitemia vera. A sensibilidade tátil e à dor pode ser prejudicada.
Na maioria dos pacientes, o baço aumenta de tamanho, o que pode se manifestar como uma sensação de peso no hipocôndrio esquerdo ou uma sensação de saciedade excessivamente rápida ao comer.
Como a doença se desenvolve? O curso da eritremia pode ser relativamente benigno, quando os pacientes vivem muitos anos sem complicações graves. Em alguns casos, poucos anos após a primeira manifestação da doença, ocorre trombose grave dos vasos cerebrais ou abdominais, levando à morte.
Uma imagem objetiva no primeiro estágio é dada por um exame de sangue. Deve-se suspeitar de eritremia se o número de glóbulos vermelhos exceder 5,7 * 1.012/l para homens e mais de 5,2 * 1.012/l para mulheres. O nível de hemoglobina é superior a 177 g/l nos homens e 172 g/l nas mulheres. O diagnóstico desta doença é feito com base em critérios especiais. Um componente necessário da busca diagnóstica é a biópsia trefina do ílio.
 A sangria alivia a condição de pacientes com policitemia.
A sangria alivia a condição de pacientes com policitemia. A policitemia vera é mais frequentemente tratada ambulatorialmente. As indicações para internação são doença grave, diminuição do número de leucócitos e plaquetas após tratamento com citostáticos, necessidade de punção de medula óssea ou baço, trepanobiópsia do ílio. O paciente deve ser hospitalizado se for planejada uma cirurgia, mesmo que pequena (por exemplo, extração dentária).
O programa de tratamento inclui as seguintes áreas:
Este é o principal tratamento para pessoas com menos de 50 anos. Quando parte do sangue circulante é removida do corpo, o leito vascular é descarregado, a coceira na pele é aliviada e a probabilidade de complicações trombóticas é reduzida.
Como método independente de tratamento, a sangria é usada para policitemia benigna, bem como durante os períodos reprodutivos e pré-menopausa. Se a eritremia recidivar após um curso de quimioterapia, a sangria também pode ser prescrita. Devem ser realizados até que haja uma diminuição significativa dos níveis de hemoglobina (não superior a 150 g/l).
O procedimento geralmente é realizado em ambiente clínico. Durante uma sessão, são retirados de 350 a 500 ml de sangue. As sessões de sangria são repetidas a cada 2 dias até que os resultados desejados sejam alcançados. No futuro, os exames de sangue serão monitorados uma vez a cada dois meses.
Como resultado da sangria, o conteúdo de ferro no corpo diminui. Na maioria das vezes, os pacientes toleram bem. No entanto, às vezes ocorrem fraqueza, perda de cabelo e anemia grave por deficiência de ferro. Neste caso, os suplementos de ferro devem ser prescritos em combinação com medicamentos citostáticos.
A sangria não é prescrita se seu efeito for pequeno e de curto prazo ou se houver sinais graves de deficiência de ferro.
Durante este procedimento, 1–1,4 litros de sangue são retirados do leito vascular do paciente. Os glóbulos vermelhos são removidos usando equipamento especial. O plasma restante é levado ao volume original com solução salina e despejado no sistema venoso. A eritrocitaférese é uma alternativa à sangria. Os cursos desse tratamento proporcionam um efeito por 1-2 anos.
Nos casos graves da doença, quando a sangria é ineficaz, em pessoas com mais de 50 anos são prescritos medicamentos citostáticos. Eles inibem a proliferação de células na medula óssea. Como resultado, o número de todas as células sanguíneas, incluindo glóbulos vermelhos, diminui. Ao tratar com citostáticos, são realizados exames de sangue regularmente para monitorar a eficácia e segurança do tratamento.
Os mais comumente usados são citostáticos alquilantes e antimetabólitos. O fósforo radioativo 32P é utilizado com menor frequência, sendo indicado principalmente para idosos.
Para trombose vascular, são prescritos antiplaquetários e heparina. Na tromboflebite aguda, é realizado tratamento local: resfriamento da perna com bolsas de gelo no primeiro dia, depois pomada de heparina e pomada Vishnevsky.
Para sangramento grave, ácido aminocapróico, plasma fresco congelado e esponja hemostática são prescritos localmente.
A eritromelalgia (dor nas pontas dos dedos, solas dos pés) é tratada (indometacina, voltaren). A heparina também pode ser prescrita.
Em caso de acidente vascular cerebral, hipertensão, úlcera gástrica, são utilizados regimes medicamentosos adequados. Eles são usados para tratar coceira na pele. A cimetidina (um bloqueador do receptor H2) às vezes é eficaz.
Indicações para remoção do baço por eritremia.
– hemoblastose crônica, que se baseia na proliferação ilimitada de toda mielopoiese, predominantemente eritrocitária. Clinicamente, a policitemia se manifesta por sintomas cerebrais (peso na cabeça, tontura, zumbido), síndrome trombohemorrágica (trombose arterial e venosa, sangramento), distúrbios microcirculatórios (frieza dos membros, eritromelalgia, hiperemia da pele e mucosas). As informações diagnósticas básicas são obtidas a partir do estudo do sangue periférico e da medula óssea. Para tratar a policitemia, são utilizadas sangrias, eritrocitaférese e quimioterapia.

O desenvolvimento da policitemia é precedido por alterações mutacionais nas células-tronco hematopoiéticas pluripotentes, que dão origem às três linhagens celulares da medula óssea. A mutação mais comum detectada é o gene da tirosina quinase JAK2 com a substituição da valina pela fenilalanina na posição 617. Às vezes há incidência familiar de eritremia, por exemplo, entre judeus, o que pode indicar uma correlação genética.
Na policitemia, existem 2 tipos de células precursoras hematopoiéticas eritróides na medula óssea: algumas delas se comportam de forma autônoma, sua proliferação não é regulada pela eritropoietina; outros, como esperado, são dependentes de eritropoietina. Acredita-se que a população autônoma de células nada mais é do que um clone mutante - principal substrato da policitemia.
Na patogênese da eritremia, o papel principal pertence ao aumento da eritropoiese, que resulta em eritrocitose absoluta, comprometimento das propriedades reológicas e de coagulação do sangue, metaplasia mieloide do baço e do fígado. A alta viscosidade do sangue causa tendência à trombose vascular e danos hipóxicos aos tecidos, e a hipervolemia causa aumento do suprimento de sangue aos órgãos internos. No final da policitemia, observa-se depleção da hematopoiese e mielofibrose.
Na hematologia, existem 2 formas de policitemia - verdadeira e relativa. A policitemia relativa se desenvolve com contagens normais de glóbulos vermelhos e diminuição do volume plasmático. Essa condição é chamada de estresse ou falsa policitemia e não é discutida no escopo deste artigo.
A policitemia vera (eritremia) pode ser de origem primária ou secundária. A forma primária é uma doença mieloproliferativa independente, baseada em danos à linhagem mieloide da hematopoiese. A policitemia secundária geralmente se desenvolve com atividade aumentada da eritropoetina; esta condição é uma reação compensatória à hipóxia geral e pode ocorrer com patologia pulmonar crônica, defeitos cardíacos “azuis”, tumores adrenais, hemoglobinopatias, ao subir em altitude ou fumar, etc.
A policitemia vera passa por 3 estágios de desenvolvimento: inicial, avançado e terminal.
Estágio I(inicial, assintomático) – dura cerca de 5 anos; é assintomático ou com manifestações clínicas minimamente expressas. Caracterizado por hipervolemia moderada, eritrocitose leve; O tamanho do baço é normal.
Estágio II(eritrêmico, expandido) é dividido em dois subestágios:
Estágio III(anêmico, posteritrêmico, terminal). Caracterizada por anemia, trombocitopenia, leucopenia, transformação mieloide do fígado e baço, mielofibrose secundária. Possíveis resultados da policitemia em outras hemoblastoses.
A eritremia se desenvolve gradualmente durante um longo período de tempo e pode ser detectada acidentalmente durante um exame de sangue. Os primeiros sintomas, como peso na cabeça, zumbido, tontura, visão turva, membros frios, distúrbios do sono, etc., são frequentemente atribuídos à idade avançada ou a doenças concomitantes.
A característica mais característica da policitemia é o desenvolvimento da síndrome pletórica, causada por pancitose e aumento do volume sanguíneo. A evidência de pletora é telangiectasia, coloração vermelho-cereja da pele (especialmente rosto, pescoço, mãos e outras áreas abertas) e membranas mucosas (lábios, língua), hiperemia da esclera. Um sinal diagnóstico típico é o sinal de Cooperman - a cor do palato duro permanece normal, mas o palato mole adquire uma tonalidade cianótica estagnada.
Outro sintoma característico da policitemia é a coceira na pele, que se intensifica após procedimentos hídricos e às vezes se torna insuportável. As manifestações específicas da policitemia também incluem eritromelalgia - uma sensação dolorosa de queimação nas pontas dos dedos, acompanhada de hiperemia.
No estágio avançado da eritremia podem ocorrer enxaquecas dolorosas, dores ósseas, cardialgia e hipertensão arterial. 80% dos pacientes apresentam esplenomegalia moderada ou grave; o fígado aumenta com menos frequência. Muitos pacientes com policitemia notam aumento do sangramento nas gengivas, hematomas na pele e sangramento prolongado após a extração dentária.
A consequência da eritropoiese ineficaz na policitemia é um aumento na síntese de ácido úrico e uma violação do metabolismo das purinas. Isto encontra expressão clínica no desenvolvimento da chamada diátese de urato - gota, urolitíase, cólica renal.
O resultado da microtrombose e da violação do trofismo da pele e das membranas mucosas são úlceras tróficas de perna, úlceras gástricas e duodenais. As complicações mais comuns na clínica de policitemia são trombose vascular de veias profundas, vasos mesentéricos, veias portais, artérias cerebrais e coronárias. Complicações trombóticas (EP, acidente vascular cerebral isquêmico, infarto do miocárdio) são as principais causas de morte em pacientes com policitemia. Ao mesmo tempo, juntamente com a formação de trombos, os pacientes com policitemia são propensos à síndrome hemorrágica com desenvolvimento de sangramento espontâneo em vários locais (gengival, nasal, das veias esofágicas, gastrointestinal, etc.).
As alterações hematológicas que caracterizam a policitemia são decisivas no diagnóstico. Um exame de sangue revela eritrocitose (até 6,5-7,5x10 12 /l), aumento da hemoglobina (até 180-240 g/l), leucocitose (acima de 12x10 9 /l), trombocitose (acima de 400x10 9 /l). A morfologia dos eritrócitos, via de regra, não se altera; com aumento do sangramento, a microcitose pode ser detectada. A confirmação confiável da eritremia é um aumento na massa de glóbulos vermelhos circulantes superior a 32-36 ml/kg. Neurologista, cardiologista, gastroenterologista, urologista.
Para normalizar o volume do Cco e reduzir o risco de complicações trombóticas, a primeira medida é a sangria. As exfusões de sangue são realizadas no volume de 200-500 ml 2 a 3 vezes por semana, seguidas de reposição do volume sanguíneo retirado com solução salina ou reopoliglucina. A consequência da sangria frequente pode ser o desenvolvimento de anemia por deficiência de ferro. A sangria para policitemia pode ser substituída com sucesso pela eritrocitoférese, que permite que apenas a massa de glóbulos vermelhos seja removida da corrente sanguínea, devolvendo o plasma.
Em caso de alterações clínicas e hematológicas pronunciadas, desenvolvimento de complicações vasculares e viscerais, recorrem à terapia mielossupressora com citostáticos (busulfan, mitobronitol, ciclofosfamida, etc.). Às vezes, é administrada terapia com fósforo radioativo. Para normalizar o estado de agregação do sangue, são prescritos heparina, ácido acetilsalicílico, dipiridamol sob o controle de um coagulograma; para hemorragias, estão indicadas transfusões de plaquetas; para diátese de urato - alopurinol.
O curso da eritremia é progressivo; a doença não é propensa a remissões espontâneas e cura espontânea. Os pacientes são forçados a ficar sob a supervisão de um hematologista por toda a vida e a passar por cursos de terapia de hemoexfusão. Na policitemia existe um alto risco de complicações tromboembólicas e hemorrágicas. A incidência de transformação da policitemia em leucemia é de 1% em pacientes que não foram submetidos a tratamento quimioterápico e de 11 a 15% naqueles que recebem terapia citotóxica.
A policitemia é uma doença que pode ser identificada apenas olhando para o rosto de uma pessoa. E se você fizer um exame diagnóstico, não haverá dúvida alguma. Na literatura médica você pode encontrar outros nomes para esta patologia: eritremia, doença de Vaquez. Independentemente do termo escolhido, a doença representa uma grave ameaça à vida humana. Neste artigo falaremos mais detalhadamente sobre o mecanismo de sua ocorrência, sintomas primários, estágios e métodos de tratamento propostos.
A policitemia vera é um câncer mieloproliferativo do sangue que produz glóbulos vermelhos em quantidades excessivas. Em menor grau, observa-se um aumento de outros elementos enzimáticos, nomeadamente leucócitos e plaquetas.
Os glóbulos vermelhos (também conhecidos como eritrócitos) saturam todas as células do corpo humano com oxigênio, transportando-o dos pulmões para os sistemas orgânicos internos. Eles também são responsáveis por remover o dióxido de carbono dos tecidos e transportá-lo até os pulmões para posterior exalação.
Os glóbulos vermelhos são produzidos continuamente na medula óssea. É um conjunto de tecidos esponjosos, localizados no interior dos ossos e responsáveis pelo processo de hematopoiese.
Os leucócitos são glóbulos brancos que ajudam a combater várias infecções. As plaquetas são fragmentos que são ativados quando a integridade dos vasos sanguíneos é perturbada. Eles têm a capacidade de grudar uns nos outros e entupir o buraco, parando assim o sangramento.
A policitemia vera é caracterizada pela produção excessiva de glóbulos vermelhos.
Essa patologia costuma ser diagnosticada em pacientes adultos, mas pode ocorrer em adolescentes e crianças. Por muito tempo a doença pode não se fazer sentir, ou seja, pode ser assintomática. Segundo estudos, a idade média dos pacientes varia de 60 a aproximadamente 79 anos. Os jovens adoecem com muito menos frequência, mas a sua doença é muito mais grave. De acordo com dados estatísticos, os representantes do sexo forte são diagnosticados com policitemia várias vezes com mais frequência.

A maioria dos problemas de saúde associados a esta doença surge do aumento contínuo do número de glóbulos vermelhos. Como resultado, o sangue fica excessivamente espesso.
Por outro lado, o seu aumento da viscosidade provoca a formação de coágulos (trombos). Eles podem interferir no fluxo sanguíneo normal através das artérias e veias. Essa situação costuma causar derrames e ataques cardíacos. O fato é que o sangue espesso flui várias vezes mais devagar pelos vasos. O coração tem que fazer mais esforços para literalmente empurrá-lo.
A diminuição do fluxo sanguíneo não permite que os órgãos internos recebam a quantidade necessária de oxigênio. Isto implica o desenvolvimento de insuficiência cardíaca, dores de cabeça, angina, fraqueza e outros problemas de saúde que não devem ser ignorados.
Infelizmente, atualmente os especialistas não conseguem dizer quais fatores levam ao desenvolvimento de uma doença como a policitemia vera.
A maioria está inclinada à teoria genética viral. Segundo ele, vírus especiais (são cerca de 15 no total) são introduzidos no corpo humano e, sob a influência de certos fatores que afetam negativamente a defesa imunológica, penetram nas células da medula óssea e dos gânglios linfáticos. Então, em vez de amadurecerem como esperado, essas células começam a se dividir e a se multiplicar rapidamente, formando cada vez mais novos fragmentos.

Por outro lado, a causa da policitemia pode estar oculta em uma predisposição hereditária. Os cientistas provaram que parentes próximos do doente, bem como pessoas com estrutura cromossômica anormal, são mais suscetíveis a esta doença.
A partir do segundo estágio de desenvolvimento da doença, literalmente todos os sistemas de órgãos internos são atraídos para o processo patológico. Abaixo listamos as sensações subjetivas do paciente.
A policitemia vera também pode ser acompanhada pelos seguintes sintomas. Em cada caso específico, sua gravidade varia.

Em primeiro lugar, o médico coleta um histórico médico completo. Ele pode fazer uma série de perguntas esclarecedoras: quando exatamente apareceu o mal-estar/falta de ar/desconforto doloroso, etc. É igualmente importante determinar a presença de doenças crônicas, maus hábitos e possíveis contatos com substâncias tóxicas.
Um exame físico é então realizado. O especialista determina a cor da pele. Por palpação e batidas, um baço ou fígado aumentado é detectado.
Para confirmar a doença, são necessários exames de sangue. Se o paciente tiver esta patologia, os resultados do teste podem ser os seguintes:
O diagnóstico também envolve aspiração cerebral e biópsia. A primeira versão do estudo envolve a retirada da parte líquida do cérebro e uma biópsia do componente sólido.
A doença policitemia é confirmada por testes de mutação genética.

Não é possível superar completamente uma doença como a policitemia vera. É por isso que a terapia se concentra exclusivamente na redução das manifestações clínicas e na redução das complicações trombóticas.
Os pacientes recebem primeiro uma sangria prescrita. Este procedimento envolve a retirada de uma pequena quantidade de sangue (de 200 a aproximadamente 400 ml) para fins terapêuticos. É necessário normalizar os parâmetros quantitativos do sangue e reduzir sua viscosidade.

Os pacientes geralmente recebem aspirina para reduzir o risco de desenvolver vários tipos de complicações trombóticas.
A quimioterapia é usada para manter um hematócrito normal quando ocorre prurido grave ou aumento da trombocitose.
O transplante de medula óssea para esta doença é realizado extremamente raramente, uma vez que esta patologia não é fatal se tratada adequadamente.
Deve-se notar que o regime de tratamento específico é selecionado individualmente em cada caso. A terapia descrita acima é apenas para fins informativos. Não é recomendado tentar lidar com esta doença sozinho.
Esta doença é bastante grave, por isso o seu tratamento não deve ser negligenciado. Caso contrário, a probabilidade de complicações desagradáveis aumenta. Isso inclui o seguinte:

A doença de Váquez é uma doença rara. Os sintomas que aparecem nos estágios iniciais de seu desenvolvimento devem ser motivo de exame imediato e terapia subsequente. Na ausência de tratamento adequado, se a doença não for diagnosticada em tempo hábil, ocorre a morte. A principal causa de morte são, na maioria das vezes, complicações vasculares ou transformação da doença em leucemia crônica. No entanto, a terapia competente e o cumprimento estrito de todas as recomendações do médico podem prolongar significativamente a vida do paciente (em 15 a 20 anos).
Esperamos que todas as informações apresentadas neste artigo sejam realmente úteis para você. Seja saudável!
Policitemia vera (eritremia, doença de Vaquez, policitemia rubra) - PV é uma doença mieloproliferativa neoplásica crônica com danos às células-tronco, proliferação de três linhagens hematopoiéticas, aumento da produção de glóbulos vermelhos e, em menor extensão, de leucócitos e plaquetas. Em determinado estágio da doença, ocorre metaplasia mieloide do baço.
A frequência da policitemia vera é de aproximadamente 1 acidente por 100 mil habitantes por ano e nos últimos anos tem tendência indiscutível de aumento. Os homens adoecem com um pouco mais de frequência do que as mulheres (1.2:1). A idade média dos pacientes é de 60 anos, os pacientes com menos de 40 anos representam apenas 5%.
Etiopatogenia. A policitemia vera é uma doença neoplásica clonal, que se baseia na transformação de uma célula-tronco hematopoiética. Como a transformação maligna ocorre ao nível de uma célula-tronco pluripotente, todas as três linhagens de hematopoiese estão envolvidas no processo. Em pacientes que sofrem de PV, há aumento do conteúdo de UFC-GEMM (unidades formadoras de colônias - granulocíticas, eritróides, macrófagos e megacariócitos) - células progenitoras próximas a uma célula-tronco pluripotente. Na cultura celular, a proliferação activa destas células ocorre na ausência de eritropoietina. Níveis séricos baixos de eritropoetina são um sinal específico de PV. Na medula óssea, observa-se hiperplasia de células predominantemente eritróides, bem como de linhagens granulocíticas e megacariocíticas. Uma característica é a presença de aglomerados de megacariócitos polimórficos (de pequenos a gigantes). A mielofibrose raramente é observada no momento do diagnóstico, mas se manifesta claramente com um longo curso da doença. Gradualmente, o número de fibras de reticulina e colágeno aumenta, a mielofibrose se desenvolve e a mielopoiese é reduzida. A massa de eritrócitos circulantes (MCE) aumenta, o hematócrito aumenta, a viscosidade do sangue aumenta (há um aumento significativo no conteúdo de hemoglobina no sangue (de 180 g/le acima), glóbulos vermelhos (de 6,6 x 10 12 /l) e hematócrito (de 55% e superior). Esses fatores, juntamente com a trombocitose, levam à microcirculação prejudicada e complicações tromboembólicas. Paralelamente, a metaplasia mieloide do baço está associada. Na IP não há marcador citogenético específico, entretanto, em um número significativo número de pacientes com PI na fase de desenvolvimento de mielofibrose, anomalias cromossômicas.
Quadro clínico muda com o curso da doença e é determinado principalmente pelo estágio da doença. Na literatura nacional, costuma-se distinguir quatro estágios da IP, que refletem os processos patológicos que ocorrem na medula óssea e no baço dos pacientes
Estágios:
I - inicial, pouco sintomático (5 anos ou mais):
o baço não é palpável
eritrocitose moderada
abundância moderada
na panmielose da medula óssea
Complicações vasculares e trombóticas são possíveis, mas não são comuns
As manifestações externas da doença são pletora, acrocianose, eritromelalgia (dor em queimação, parestesia nas pontas dos dedos) e coceira na pele após a lavagem. Um aumento no MCE e, consequentemente, no volume sanguíneo circulante leva à hipertensão arterial. Se o paciente já sofreu de hipertensão, o nível de pressão arterial aumenta e a terapia anti-hipertensiva torna-se ineficaz. As manifestações de doença coronariana e aterosclerose cerebral são agravadas. Como o MCE aumenta gradualmente, a pletora, um aumento no número de glóbulos vermelhos e hemoglobina, sinais de distúrbio da microcirculação em vários pacientes aparecem 2 a 4 anos antes do diagnóstico ser feito.
II – eritrêmico expandido (10-15 anos):
A. Sem metaplasia mieloide do baço
estado geral é perturbado
abundância pronunciada (Hb 200 g/l ou mais)
complicações trombóticas (acidente vascular cerebral, infarto do miocárdio, necrose das pontas dos dedos)
panmielose
eritromelalgia (dor nos membros e ossos)
No quadro do sangue periférico, além da eritrocitose, a neutrofilia costuma estar presente com desvio da fórmula leucocitária para a esquerda para mielócitos únicos, além de basofilia e trombocitose. Na medula óssea, é detectada hiperplasia total de três linhas com megacariocitose pronunciada e é possível mielofibrose reticulina. Mas nesta fase da doença ainda não há metaplasia mieloide do baço (MMS), e a esplenomegalia observada é devida ao aumento do sequestro de eritrócitos e plaquetas. As complicações vasculares são mais frequentes e graves do que na primeira fase da doença. Na patogênese da trombose, um papel importante é desempenhado pelo aumento do MCE, levando ao aumento da viscosidade sanguínea e à desaceleração do fluxo sanguíneo, trombocitose, bem como disfunção do endotélio. A isquemia associada ao fluxo sanguíneo arterial prejudicado é observada em 24-43% dos pacientes. Predomina a trombose de vasos cerebrais, coronárias e artérias que fornecem sangue aos órgãos abdominais. A trombose venosa é detectada em 25-30% dos pacientes e é a causa de morte em aproximadamente um terço dos pacientes que sofrem de PV. A trombose das veias do sistema porta e das veias mesentéricas não é incomum. Em vários pacientes, são as complicações trombóticas que se tornam uma manifestação de IP. A policitemia vera pode ser acompanhada por síndrome hemorrágica: sangramentos nasais frequentes e sangramento após extração dentária. A hipocoagulação baseia-se na desaceleração da conversão do fibrinogênio em fibrina, que ocorre proporcionalmente ao aumento do hematócrito, e na violação da retração do coágulo sanguíneo. Erosão e úlceras do estômago e duodeno são consideradas complicações viscerais da PI.
B. Com metaplasia mieloide do baço (MMS).
hepatoesplenomegalia
abundância é moderadamente expressa
panmielose
aumento de sangramento
complicações trombóticas
A esplenomegalia aumenta, o número de leucócitos aumenta, o deslocamento da fórmula leucocitária para a esquerda torna-se mais pronunciado. Na medula óssea - panmielose; A mielofibrose de reticulina e colágeno focal se desenvolve gradualmente. O número de glóbulos vermelhos e plaquetas diminui um pouco devido ao aumento da destruição no baço, bem como à substituição gradual do tecido hematopoiético por tecido fibroso. Nesta fase, pode-se observar a estabilização do quadro do paciente, o nível de hemoglobina, glóbulos vermelhos e plaquetas aproxima-se do normal sem medidas terapêuticas.
III – anêmico:
sm anêmico (até mesmo pancitopenia)
mielofibrose grave
fígado, baço aumentado
Na medula óssea, a mielofibrose do colágeno aumenta e a mielopoiese diminui. O hemograma mostra anemia, trombocitopenia e pancitopenia. O quadro clínico da doença pode incluir síndromes anêmicas e hemorrágicas, esplenomegalia e aumento da caquexia. O desfecho da doença pode ser a transformação em leucemia aguda e síndrome mielodisplásica (SMD).
Diagnóstico. Atualmente, os critérios desenvolvidos pelo American Policitemia Vera Study Group (PVSG) são utilizados para estabelecer o diagnóstico de policitemia vera. Você-
1) aumento da massa de glóbulos vermelhos circulantes (mais de 36 ml/kg para homens e mais de 32 ml/kg para mulheres);
2) saturação normal do sangue arterial com oxigênio (pO2 superior a 92%);
3) esplenomegalia.
1) trombocitose (contagem de plaquetas superior a 400 x 10 9 /l);
2) leucocitose (o número de leucócitos é superior a 12 x 10 9 /lb sem sinais de infecção);
3) atividade da fosfatase alcalina (neutrófilos acima de 100 unidades na ausência de febre ou infecção);
4) alto teor de vitamina B12 (mais de 900 pg/ml).
O diagnóstico de IP é considerado confiável se o paciente apresentar todos os três sinais da categoria A, ou se o primeiro e o segundo sinais da categoria A e quaisquer dois sinais da categoria B estiverem presentes.
Atualmente, o sinal diagnóstico mais importante é o quadro histológico característico da medula óssea; hiperplasia de células de linhagens eritróides, granulocíticas e megacariócitos com predomínio de eritróides, acúmulos de megacariócitos polimórficos (de pequenos a gigantes). A mielofibrose raramente é observada no momento do diagnóstico, mas torna-se distinta com o longo curso da doença.
No estágio I, a policitemia vera, caracterizada por eritrocitose isolada, deve ser diferenciada da eritrocitose secundária, que é uma resposta a qualquer processo patológico do organismo e pode ser verdadeira ou relativa.
A eritrocitose relativa é consequência da hemoconcentração, ou seja, o MCE é normal, mas o volume plasmático está reduzido, o que é observado com desidratação do corpo (por exemplo, uso de diuréticos, poliúria em pacientes com diabetes, vômitos e diarreia), perda de uma grande quantidade de plasma devido a queimaduras.
A verdadeira eritrocitose secundária (MCE está aumentada, o hematócrito está aumentado) é causada pelo aumento da produção de eritropoetina. Este último é de natureza compensatória e é causado pela hipóxia tecidual em pessoas que vivem em altitudes significativas acima do nível do mar, em pacientes com patologias dos sistemas cardiovascular e respiratório e em fumantes. Esta categoria também inclui pacientes com hemoglobinopatias hereditárias, caracterizadas por maior afinidade da hemoglobina pelo oxigênio, do qual uma quantidade menor é liberada nos tecidos do corpo. A produção inadequada de eritropoietina é observada em doenças renais (hidronefrose, patologia vascular, cistos, tumores, anomalias congênitas), câncer hepatocelular, grandes miomas uterinos. Uma característica diagnóstica diferencial essencial é o nível de eritropoetina sérica.
Tratamento. Nos estágios iniciais da doença, recomenda-se a utilização de sangria, o que ameniza significativamente as manifestações da síndrome pletórica. O método de escolha para redução do hematócrito (e da hemoglobina para valores normais) é a sangria (exfusão), recomendada se o hematócrito ultrapassar 0,54. O objetivo do tratamento é um hematócrito inferior a 0,42 para mulheres e 0,45 para homens.Nas condições modernas, a sangria pode ser substituída pela eritrocitoférese. Além disso, para facilitar a sangria e prevenir complicações trombóticas, os pacientes recebem cursos de terapia desagregante (aspirina, reopoliglucina, etc.). A escolha de um método de tratamento no estágio II avançado da PI talvez seja a tarefa mais difícil. Além da eritrocitose, os pacientes apresentam leucocitose e trombocitose, podendo esta última atingir números muito elevados. Alguns pacientes já sofreram algum tipo de complicação trombótica e as exfusões aumentam o risco de trombose.
Ao individualizar a terapia, a idade dos pacientes deve ser levada em consideração. É o tratamento de pacientes com menos de 50 anos, sem histórico de complicações trombóticas e hipertrombocitose grave (< 1000,0 х 10 9 /л) может быть ограничено только кровопусканиями в сочетании с терапией аспирином (или без него) в дозе 100-375 мг в день.
Para pacientes com mais de 70 anos, com história de complicações trombóticas e hipertrombocitose grave, está indicada terapia com drogas mielossupressoras. Pacientes entre 50 e 70 anos de idade sem complicações trombóticas ou hipertrombocitose grave podem ser tratados com agentes mielossupressores ou flebotomia, embora este último tratamento possa aumentar o risco de complicações trombóticas.
Atualmente, além dos agentes sangüíneos e antiplaquetários, a hidroxiureia e o alfa-interferon são usados principalmente para o tratamento da PV, menos frequentemente o busulfan, e a anagrelida é usada no exterior. A hidroxiureia pode ser a droga de escolha em pacientes com PV com leucocitose e trombocitose graves. Mas para pacientes jovens, o uso da hidroxiureia é limitado pelos seus efeitos mutagênicos e leucosogênicos. Além da hidroxiureia, o interferon-alfa é amplamente utilizado no tratamento da PV. Em primeiro lugar, o IF-a suprime muito bem a proliferação patológica e não tem efeito leucêmico. Em segundo lugar, tal como a hidroxiureia, reduz significativamente a produção de plaquetas e glóbulos brancos. Particularmente digna de nota é a capacidade do IF-a de eliminar a coceira na pele causada por procedimentos com água.
A aspirina na dose diária de 50-250 mg, via de regra, elimina distúrbios da microcirculação. O uso deste medicamento ou de outros agentes antiplaquetários para fins terapêuticos ou profiláticos é recomendado para todos os pacientes com IP.
Infelizmente, atualmente não existe tratamento eficaz para PV anêmica em estágio III. A terapia é limitada a paliativos. A síndrome anêmica e hemorrágica é corrigida por transfusões de hemocomponentes. Foi relatada a eficácia do transplante de células-tronco hematopoiéticas em pacientes com PV no estágio de mielofibrose com esplenomegalia e pancitopenia e transformação em leucemia aguda ou SMD. A taxa de sobrevivência de três anos dos pacientes após o transplante foi de 64%.
Previsão. Apesar de seu curso longo e em alguns casos favorável, a PI é uma doença grave e repleta de complicações fatais que encurtam a expectativa de vida dos pacientes. A causa mais comum de morte em pacientes é trombose e embolia (30-40%). Em 20-50% dos pacientes no estágio de mielofibrose pós-policitêmica (PV estágio III), ocorre a transformação em leucemia aguda, que tem um prognóstico desfavorável - uma taxa de sobrevida em três anos de apenas 30%.

Em 2018, o Jejum da Natividade começará em 28 de novembro. Durante este período, os crentes ortodoxos se preparam para celebrar o Natal...

Constituir família é o sonho da maioria das mulheres. Eles querem ter um marido amoroso e um monte de filhos. Mas nem sempre é um relacionamento...

Este artigo contém: a oração mais poderosa pelo divórcio - informações retiradas de todo o mundo, da rede eletrônica e...

Site de informações sobre ícones, orações, tradições ortodoxas. Oração pelos escândalos e brigas na família, com o marido, com os filhos...

O que seria do Ano Novo sem champanhe, tangerinas, Olivier, formol e o “arenque com casaco de pele” preferido de todos. Com o último...

Vamos preparar os ingredientes necessários para os biscoitos. A primeira coisa a fazer é colocar a água para ferver. Nós precisamos...

É possível cadastrar funcionário para o cargo de diretor financeiro - contador-chefe? O contador-chefe afirma...

O chefe de uma pequena empresa pode gerenciar facilmente o orçamento de forma independente. VERIFICADO! Se você conseguir...

A criação de novos projetos envolve uma justificação económica preliminar para a sua viabilidade, posterior...
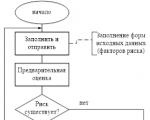
O reporte é gerado pelo RM, é acordado (aprovado) pelo Comité de Risco no âmbito do Conselho de Administração e transmitido a...

Na beira de um prado grande, muito grande, em uma longa folha de grama esmeralda vivia uma pequena joaninha. O pequeno de Deus...

Hoje em dia é bastante comum as pessoas se voltarem para as estrelas. Com a ajuda de um horóscopo uma pessoa pode descobrir...

Negócios ou amigável. Se você estava perseguindo um estranho, isso significa o seu nível de confiança no sexo feminino...

de acordo com o livro dos sonhos de Freud Se você sonhou que estava pescando, isso significa que na vida real dificilmente você consegue desligar...

Constituir família é o sonho da maioria das mulheres. Eles querem ter um marido amoroso e um monte de filhos. Mas nem sempre é...

Este artigo contém: a oração mais poderosa pelo divórcio - informações retiradas de todo o mundo, eletrônicas...