Філософське бачення проблеми
Сенс життя пов'язаний із питанням «Заради чого жити», а не питанням про те, як підтримувати життя. Ставлення людини як...
З розвитком молекулярно-генетичних методів процес діагностики різних захворюваньсильно спростився. Синдром Прадера - Віллі - вроджене захворюванняспадкової природи, що супроводжується великою кількістю ускладнень. Патологія є частою причиноюожиріння в дітей віком.
Синдром Прадера – Віллі є генетичним захворюванням, що виникає через порушення функціонування певного району 15-ї хромосоми, успадкованої від батька. Гени у ньому можуть частково не працювати, бути віддаленими чи нести лише матеріал, отриманий матері.
У наш час захворювання дуже часто неправильно діагностується, у зв'язку з чим у перші роки життя дитина не отримує адекватного та своєчасного лікування.
Вперше синдром був описаний швейцарськими педіатрами А. Прадером та Г. Віллі. За статистикою, захворювання зустрічається в одного з 10–20 тисяч новонароджених. Довгий часбуло складно встановити зв'язок між спадковістю та причиною виникнення патології. З розвитком науки та появою різних молекулярних методів тестування вчені змогли точно ідентифікувати генетичне захворюванняу всіх пацієнтів.
Існувало багато ймовірних варіантів передачі захворювання. За допомогою генетичних аналізів було встановлено, що до порушення призводить до 70% випадків мікроделеція (випадання ділянки хромосоми) або відсутність активності генів ділянки 15 хромосоми, отриманої від батька, а у 25% пацієнтів - материнська ізодісомія (всі дві хромосоми отримані від матері). Частина, що залишилася, виникає через заміну одних генів на інші в певній ділянці 15 хромосоми або порушення їх функціонування (імпринтінгу).
Імпринтинг визначає ступінь впливу генів на будову та функціонування організму залежно від батька, від якого вони були отримані. Подібна ситуаціяхарактерна не для всього спадкового матеріалу, а лише для 1 відсотка.
Клінічних відмінностей між хворими у своїй був виявлено.
Існує кілька видів порушень, при яких виникає синдром Прадера – Віллі у дітей різного віку
Захворювання зустрічається однаково часто серед хлопчиків та дівчаток. Синдром Прадера - Віллі є головною причиноюрозвитку ожиріння в дітей віком до року. У хворих знижений процес розщеплення жиру та посилено його вироблення.
Діти народжуються доношеними. За зовнішніми даними буває складно визначити одразу наявність генетичного захворювання. Однак при детальному вивченні виявляється дуже багато відхилень.
У дітей до півтора року знижений м'язовий тонус, реакція у вигляді скидання рук при переляку (рефлекс Моро) та сосально-ковтальні рефлекси. Батьки мають сильні проблеми при годівлі дитини. Часто зустрічається утруднене дихання.
Рефлекс Моро проявляється у нормі в дітей віком із народження і пропадає за кілька місяців. Під час огляду лікар ляскає поблизу немовляти і спостерігає за ним. У нормі дитина сильно розводить ручки та розчепірює пальці, після чого починає швидко їх зближувати. Збоку здається, що малюк намагається обійняти і притиснути до себе когось.
 У перші півтора роки життя у дітей із синдромом Прадера – Віллі яскраво виражено зниження м'язового тонусу
У перші півтора роки життя у дітей із синдромом Прадера – Віллі яскраво виражено зниження м'язового тонусу
Пацієнти з синдромом Прадера – Віллі страждають від дисфункції центрального органу, що управляє всією ендокринною системою - гіпоталамуса, розташованого в головному мозку Недорозвиток яєчок і яєчників, що виникає внаслідок цього, призводить до порушення вироблення гормонів і недорозвинення зовнішніх і внутрішніх статевих ознак. У хлопчиків цей процес є причиною відсутності одного або обох яєчок у мошонці, а у дівчаток у половині випадків відзначається зменшена недорозвинена матка. Зниження кількості специфічного білка-ферменту тирозинази призводить до слабкого пігментування волосся, шкіри та райдужки ока.
Крипторхізм - неправильне розташування яєчок, при якому вони не виходять у мошонку, а залишаються всередині черевної порожнинипід шкірою на стегні, лобку, в паху.
Надалі у дитини розвивається постійне почуття голоду, їжа не приносить насичення. Саме з цього моменту починається розвиток ожиріння.Для цього захворювання характерно відкладення жиру в нижній частині тулуба, в ділянці живота та стегон.
 У зв'язку з посиленим апетитом пацієнти швидко набирають вагу
У зв'язку з посиленим апетитом пацієнти швидко набирають вагу
Поступово м'язовий тонус у дитини відновлюється та розвивається до нормального рівняприблизно до шести років. Однак стають сильно помітними зовнішні дефекти: стопи і кисті рук непропорційно маленькі, ноги викривлені, надмірно опущені повіки, деформований хребет, зростання нижче середніх показників для цього віку. Дитина швидко втомлюється, часто перебуває у сонливому стані, відстає фізично від своїх однолітків. Часто розвивається через м'язові дефекти.
Психічний стан хворих часто нестабільний. Незважаючи на загальну дружелюбність, у них спостерігаються спалахи гніву, невмотивованої агресії, істерики. Діти з синдромом шкідливі, упираються. У багатьох хворих зустрічається невроз нав'язливих станів, прояви якого дуже різноманітні, починаючи з виконання побутових справ та закінчуючи страхами та фобіями. Деякі діти страждають на дерматиломанію - здирання шматочків шкіри на своєму тілі. До 10% хворих відчувають галюцинації, параноїдальні відхилення та страждають від депресій.
У шкільному віціособливо виділяється порушення мови та зниження інтелектуального розвитку.
У 1992 році вчені провели вивчення інтелекту хворих із синдромом Прадера – Віллі. У нормі коефіцієнт інтелектуального розвитку становить 85-115 балів. Лише 5% пацієнтів подолали цей поріг та залишилися на низькому або середньому рівні розвитку, решта перебувала нижче 85 балів. Більшість мало помірно знижений інтелект і 30% мали розумову відсталість.
Діти з цим синдромом мають незвичайне сприйняття, у них сильно розвинена уява, вони відмінно сприймають інформацію візуально, добре запам'ятовують слова, швидко читають, але при всьому цьому мають проблеми з усним виразом думки і мовлення. Сприймати інформацію на слух хворі можуть погано, їхнє логічне та математичне мислення перебуває на вкрай низькому рівні. Письмове вираження думок може даватися їм з великими труднощами.
У дорослого життяхворі стикаються з безліччю проблем. Переважна більшість не може мати дітей, недорозвинення статевих залоз призводить до безплідності та різних аномалій. анатомічної будовирепродуктивних органів Багатьом ставиться діагноз на цукровий діабет. У зовнішньому виглядіхворих виділяється широкий ніс, мигдалеподібні очі, вузькі губи. Колір очей, волосся та шкіри таких хворих світліший, ніж у найближчих родичів.
Одним із страшних захворювань, яке найчастіше проявляється у хворих із синдромом Прадера – Віллі, є збочене кровотворення (лейкемія). Рівень відновлення генетичної інформації у білих клітинах крові (лімфоцитах) знижено до 67%. У зв'язку з цим у хворих надзвичайно підвищений ризик онкологічних патологійсистеми кровотворення.
Проблеми з кістковою тканиноюпроявляються у всьому організмі. Зуби сильно схильні до карієсу, а сколіоз прогресує з віком. При цьому люди з цим синдромом дуже гнучкі, суглоби легко гнуться в різні боки, не викликаючи хворобливих відчуттів.
Перші ознаки захворювання лікар може виявити вже під час вагітності: слабка рухливість плода, багатоводдя, неправильні типи передлежання (ягідне, поперечне). Рекомендовано консультацію лікаря, який призначить генетичний аналіз. Для отримання клітин майбутньої дитини використовується прокол плодового міхура(Амніоцентез). При діагностиці крові матері виявляється знижений рівень гормону гонадотропіну.
Після народження самим точним визначеннямНаявність синдрому є виявлення аномалій 15 хромосоми шляхом молекулярно-генетичних досліджень.
 Існує безліч зовнішніх ознаксиндрому Прадера - Віллі: укорочені стопи та кисті, сколіоз, викривлення ніг
Існує безліч зовнішніх ознаксиндрому Прадера - Віллі: укорочені стопи та кисті, сколіоз, викривлення ніг  Вже в юному віці організм дитини посилено накопичує жирові відкладення.
Вже в юному віці організм дитини посилено накопичує жирові відкладення.  Особливо сильно страждають від ожиріння нижні кінцівки
Особливо сильно страждають від ожиріння нижні кінцівки
З 1993 року було виділено два види ознак захворювання: основні та додаткові.
Основні ознаки:
Додаткові ознаки:
При синдромі Прадера-Віллі необхідно проведення диференціальної діагностики з наступними захворюваннями:

Із завданням диференціальної діагностики повністю справляється аналіз ДНК, без якого багато випадків синдрому Прадера – Віллі залишаються невиявленими. Лікар може поставити діагноз синдром Дауна, оскільки він більш поширений та вивчений. В обох випадках спостерігаються ожиріння та розумова відсталість.
На жаль, захворювання є невиліковним і супроводжуватиме хворого все життя.Однак при ранній діагностиці та своєчасному початку терапії можна суттєво покращити життя дитини та знизити вираженість клінічних проявів.
У багатьох випадках хворим призначається прийом гормону росту у зв'язку зі зниженим виробництвом його в організмі дитини. Терапія допомагає посилити зростання пацієнта та набір м'язової масиі певною мірою навіть знизить апетит.
У зв'язку з недорозвиненням яєчок та яєчників лікар призначає гормони для стимуляції статевого розвитку. У разі крипторхізму призначають операцію для виведення яєчок у мошонку.
Так як більшість хворих страждають на порушення сну і раптовою зупинкою дихання в певні моменти, лікарем рекомендується використання спеціального медичного апарату. Пристрій запобігає такому негативному стану, створюючи постійну вентиляцію легень за рахунок тиску.
 Спеціальний пристрій допоможе уникнути зупинки дихання уві сні
Спеціальний пристрій допоможе уникнути зупинки дихання уві сні Для лікування порушень обміну речовин призначають метаболічні препарати.
З дитинства хворій дитині необхідно проходити сеанси фізіотерапії та масажу для зміцнення м'язів, підтримки їх у тонусі.
 Діти з синдромом Прадера-Віллі дуже гнучкі і гімнастика може підійти багатьом з них
Діти з синдромом Прадера-Віллі дуже гнучкі і гімнастика може підійти багатьом з них
Додаткові спортивні кружки чудово допоможуть дитині тримати м'язи в тонусі, контролювати вагу та повноцінно фізично розвиватися. Дитині підійде дитячий фітнес, ЛФК та інші секції.
У зв'язку з тим, що основна проблема хворих із синдромом Прадера-Віллі – ожиріння, лікарі приділяють посилену увагу суворій дієті. Варто обмежувати жирну їжуі швидкі вуглеводи. У дошкільному віці допускається повноцінний раціону розвиток організму. Потім призначають дієту з дефіцитом калорій (до 1200 на день), додатково використовується прийом полівітамінів та мінералів.
У разі наявності сильних проблемз годуванням у новонароджених доводиться вдаватися до харчування через шлунковий зонд.
Так як апетит у таких дітей неконтрольований, вони часто крадуть та ховають їжу. Батьки повинні зменшити доступ до їжі, використовувати різні замикаючі засоби на холодильник та шафи.
Необхідно виключити із харчування борошняну продукцію, солодощі, жирну їжу. Раціон дитини має бути насичений овочами, фруктами, нежирним м'ясом та рибою. Необхідно використовувати для приготування страв рослинні олії- оливкова, грецька, лляна.
Обов'язковим заходом при синдромі Прадера – Віллі є спостереження невропатолога, особливо на перших роках життя. Лікар точно встановить ступінь затримки розвитку та допоможе знизити вираженість симптомів хвороби.
Обов'язковим є відвідування логопеда-дефектолога. Кваліфікований лікар допоможе структурувати мовлення, навчити правильну вимову звуків.
 Заняття з логопедом необхідні дитині із синдромом Прадера - Віллі
Заняття з логопедом необхідні дитині із синдромом Прадера - Віллі Педіатр-ендокринолог має постійно перевіряти гормональний фондитини, вчасно коригувати прийом препаратів та стежити за дієтою.
Повністю вилікуватися від синдрому Прадера-Віллі неможливо, проте при правильному проведеннітерапії якість життя хворих суттєво покращується.
Тривалість життя хворих може досягати шістдесяти та більше років. Найчастіше летальні випадки трапляються наслідками ожиріння: діабету, зупинки дихання уві сні, захворювань нирок та серцево-судинної системи.
Істотно зростає тривалість життя у хворих із синдромом Прадера - Віллі в сім'ях, які дотримуються принципів правильного харчування та підтримують рухливий спосіб життя.
Так як синдром є вродженим, його профілактика може полягати тільки в допологовому аналізі спадкового матеріалу клітин плода і подальшої консультації лікаря-генетика.
Якщо у першої дитини діагностовано успадкування обох хромосом від батька або випадання в одній з них певної ділянки, то ймовірність народження наступного малюка з аналогічною патологією становить один відсоток. Однак якщо причиною синдрому Прадера – Віллі було заміщення ділянки хромосоми іншим генетичним набором, то ймовірність збільшується до 50%. Аналогічним випадком є успадкування обох хромосом від матері. Знаючи результати аналізів, можна розумно оцінити все можливі ризикинародження дитини з цим захворюванням.
 Генетичний аналіз клітин плода – метод профілактики синдрому Прадера – Віллі.
Генетичний аналіз клітин плода – метод профілактики синдрому Прадера – Віллі. Незважаючи на невиліковність хвороби, своєчасне та якісне лікуваннядопоможе дитині підтримувати нормальна вага, рухливість та розвиватися інтелектуально за допомогою спеціальних методик.
Синдромом Прадера-Віллі в медицині називають рідкісне спадкове захворюваннядля якого характерна відсутність або недостатнє функціонування деяких генів або їх частин 15-ої батьківської хромосоми. Вперше дана патологіябула описана в 1956 в Швейцарії педіатрами А. Прадером і Х. Віллі, на прізвища яких і названий синдром. Частота його становить 1 випадок на 12-15 тисяч новонароджених дітей. Симптоми та прояви синдрому Прадера-Віллі бувають різними, а перебіг захворювання залежить, як правило, від конкретного випадку.
Деякі ознаки синдрому Прадер-Віллі можна виявити ще на етапі вагітності. Насамперед, це низька рухливістьплоду та його неправильне положення. Вже після народження виявляється м'язова гіпотонія, яка зберігається протягом першого року життя малюка. Крім того, у дітей з синдромом проявляється зниження ковтального та смоктального рефлексів, що ускладнює процес годування. Порушення розвитку рухових функційу них також обумовлено м'язовою гіпотонією, тому хворим дітям буває важко сидіти, тримати голову тощо. Однак важливо відзначити, що гіпотонія зменшується і до віку 6-7 років практично зникає.
Синдром Прадера-Віллі у дітей також проявляється постійним почуттямголоду та відсутністю насичення. Ця ознакаЗахворювання зазвичай виникає на другому-четвертому році життя дитини. На цьому тлі поступово розвивається гіперфагія або ненажерливість, нав'язливі думки про їжу та обсесивну поведінку, яка спрямована на безперервні пошуки їжі та задоволення почуття голоду. Такі симптоми неминуче призводять до ожиріння, яке у разі даного захворюванняспостерігається в основному на тулубі та проксимальних відділах кінцівок. Ці ознаки синдрому Прадера-Віллі у дітей нерідко призводить до такого ускладнення, як обструктивне апное, що виявляється зупинкою дихання уві сні.
Іншими типовими симптомамизахворювання виступають:
Вже при народженні синдром Прадер-Віллі у дітей проявляється порушенням розвитку статевих органів. У хлопчиків з цим захворюванням спостерігається недорозвинення статевого члена та мошонки, а рівень тестостерону різко знижений, у дівчаток – недорозвинення статевих губ та нерідко матки. Надалі хвороба призводить до відсутності або затримки статевого дозрівання та безпліддя.
Однією з основних ознак синдрому Прадера-Віллі є також затримка психомоторного розвитку. Коефіцієнт інтелектуального розвитку у хворих становить 20-80 одиниць, тоді як нормою є показник 85-115 одиниць. Разом з цим діти, які страждають на дане захворювання, як правило, мають хорошу зорову пам'ять, можуть навчитися читати і навіть мати досить багатий пасивний словник, проте їх мова значно гірша, ніж розуміння.
Синдром Прадера-Віллі у дітей зазвичай супроводжується поганою слуховою та зоровою пам'яттю, математичні навички та навички листа даються їм дуже важко. Слід зазначити, що нерідко у дітей із таким синдромом розвивається цукровий діабет.
Рання діагностика синдрому Прадера-Віллі та подальше лікування дозволяють покращити прогноз розвитку захворювання. Діагноз ставиться, як правило, на основі клінічних проявів хвороби, але на сьогодні часто використовується генетичне тестування, яке фахівці рекомендують насамперед для новонароджених. Це зумовлено тим фактом, що у дітей наявність синдрому визначити набагато важче, оскільки неможливо перевірити їх здібності, що дозволяють проводити діагностику синдрому Прадера-Віллі за клінічними проявами.
Генетичне тестування проводиться методом ДНК-метилювання з метою з'ясування, чи є присутніми на 15 хромосомі відхилення, що призводять до виникнення захворювання. Цей спосібДіагностика синдрому Прадера-Віллі допомагає виявити 97% випадків хвороби.
Варто також зазначити, що часто захворювання діагностується неправильно, оскільки його нерідко плутають із синдромом Дауна, який зустрічається набагато частіше. До того ж такий характерна ознакасиндром Прадера-Віллі, як ожиріння, може бути присутнім також при синдромі Дауна. Тому величезна кількість випадків хвороби залишаються не виявленими.
 Оскільки хвороба є генетичною аномалією, для лікування синдрому Прадера-Віллі на сьогоднішній день не існує жодних ефективних лікарських засобів. Разом з цим застосовуються деякі лікувальні заходи, які допомагають покращити якість життя хворих. Насамперед вони мають бути спрямовані на підвищення м'язового тонусу, тому хворі діти потребують спеціальних масажних процедур та фізіотерапії.
Оскільки хвороба є генетичною аномалією, для лікування синдрому Прадера-Віллі на сьогоднішній день не існує жодних ефективних лікарських засобів. Разом з цим застосовуються деякі лікувальні заходи, які допомагають покращити якість життя хворих. Насамперед вони мають бути спрямовані на підвищення м'язового тонусу, тому хворі діти потребують спеціальних масажних процедур та фізіотерапії.
Лікування синдрому Прадера-Віллі включає також дієти з обмеженням жирів та вуглеводів. Щоб уникнути ожиріння, необхідно постійно стежити за якістю та калорійністю їжі, що вживається. Крім того, при лікуванні нерідко рекомендується застосування гормональної терапіїза допомогою гонадотропінів, які дозволяють збільшити ріст хворої дитини та відновити тонус м'язів. Це сприяє правильному розподілу калорій в організмі, перешкоджаючи ожирінню.
Лікування синдрому Прадера-Віллі також передбачає спеціальні методики розвитку хворих дітей, заняття з дефектологом, логопедом та психологом.
Відео з YouTube на тему статті:
Синдром Прадера-Віллі – рідкісна генетична проблема, що характеризується втратою батьківської 15 хромосоми. Подібний дефект супроводжується розвитком ознак гіпогонадизму, ожиріння та розумової відсталості. Перші симптоми захворювання виявляються ще в дитячому віці, найчастіше посилюючись у міру зростання та розвитку дитини. Діагностика патології ґрунтується на оцінці функції ендокринної системиу поєднанні зі специфічними ознаками розладу. Лікування має симптоматичний характер і спрямоване на зниження інтенсивності проявів хвороби, а також профілактику виникнення ускладнень.
Перша згадка про патологію датується 1887 роком. Ленгдон Даун описав дівчинку-підлітка, у якої відзначалася затримка фізичного розвитку, гіпогонадизм та ожиріння. Спочатку хвороба отримала назву полісарція. Повноцінну характеристику синдрому дали швейцарські лікарі Прадер, Віллі та Лабхарт у 1956 році. Пізніше в ході глибокого вивчення лікарі визначили і точну локалізацію генетичної мутації, яка призводила до виникнення захворювання у дітей. Вони також пов'язали зміни із синдромом Ангельмана. Обидва розлади провокуються дефектом у будові 15 хромосоми. При цьому в одному випадку аномалія формується в материнській копії, а в іншому – батьківській. Патологія була названа синдромом Віллі-Прадера на честь лікарів, які зробили найбільший внесок у її вивчення. Хвороба відноситься до рідкісних, так як її поширеність коливається в межах одного випадку на 10-25 тисяч новонароджених. Статевої або расової схильності не встановлено.
У генетиці прийнято диференціювати кілька дефектів каріотипу, що призводять до розвитку синдрому Прадер-Віллі. Вони зумовлюють інтенсивність прояву симптомів захворювання. Розрізняють такі форми:
Симптоми синдрому Прадера-Віллі реєструються ще під час вагітності. Непрямими ознакамирозвитку патології вважають малу активність плода та його неправильне розташування. Відзначається також багатоводдя та зміна рівня гонадотропіну у майбутньої матері. Подальші прояви синдрому залежить від віку пацієнта.
Вже в перші місяці після народження захворювання дається взнаки. Малюки страждають від вираженої гіпотоніїм'язів, що часто діагностується вивих стегна на тлі уродженої дисплазіїсуглоба. У дітей із синдромом Прадера-Віллі відзначають також зниження смоктального та ковтального рефлексу, аж до їх повної відсутності. Протягом кількох місяців здатність пити грудне молокоможе спонтанно відновлюватись. Пацієнти із захворюванням мають різні деформаціїособи та кінцівок, що включають мікроцефалію, недорозвиненість вушних хрящів, а також непропорційно зменшені стопи та кисті. Характерними рисами синдрому Прадера-Віллі вважається і гіпогонадизм, особливо помітний у хлопчиків. Пацієнти найчастіше крипторхи, у них недорозвинені мошонка та статевий член. Дівчатка також страждають від зниження функції статевих залоз, однак ці ознаки рідко помітні до підліткового віку. У міру розвитку дитини стають очевидними інтелектуальні відхилення, що виявляються поганою навченістю, малим словниковим запасом та іншими мовними порушеннями. У тяжких випадках пацієнти також страждають від неврологічного дефіциту, у них спостерігаються симптоми збоїв у роботі серця та респіраторної системи.
Найбільшої інтенсивності клінічні прояви синдрому Віллі-Прадера досягають пубертатному періоду. Це з вираженими відмінностями хворих від однолітків, які проходять етап статевого дозрівання. Підлітки з патологією відстають у розвитку, а також страждають на виражене ожиріння. Симптоми гіпогонадизму посилюються. У дівчаток відкладається настання менархе - першої менструації, аж до її повної відсутності, не збільшується груди. Хлопчики мають женоподібну фігуру. Зростання дітей залишається нижчим за середнє. Інтелектуальні здібностіхворих знижено, проте зберігається вміння читати та писати. Словниковий запаспоступово збільшується, хоча діти все ще мають труднощі з вербальним думкою. Підлітки страждають від підвищеної тривожностіта нервової збудливості. Подібні особливості поведінки у поєднанні зі специфічною зовнішністю призводять до виникнення труднощів у процесі соціалізації дітей.
У ряді випадків у пацієнтів реєструються важкі наслідкирозвитку синдрому Прадера-Віллі Малюки із захворюванням можуть страждати від вроджених вадсерця, які несуть загрозу їх життю та здоров'ю. Неврологічний дефіцит пов'язаний з розвитком судом, які потребують адекватного контролю, а в ряді випадків і госпіталізації дитини до спеціалізованих медичні центри. Поширені епізоди діагностики у пацієнтів цукрового діабету, що асоціюється з ожирінням, що формується на тлі метаболічних порушень. Надмірна вага негативно позначається і на стані опорно-рухового апарату. Діти посилюються деформації хребта, вони страждають від болю внаслідок неадекватного навантаження на суглоби. Пацієнти схильні до розвитку онкологічних процесів. Однак синдром Прадера-Віллі при адекватному лікуванні суттєво не впливає на тривалість життя людини.
Патологія має генетичну природу, тобто пов'язана із виникненням мутацій у хромосомному наборі людини. Розвиток специфічних клінічних ознаквикликано порушенням функції фрагментів ДНК, оскільки батьківська інформація відсутня. В результаті таких змін відзначається збій у формуванні статевих залоз. У процесі зростання та розвитку плоду відбувається прояв наслідків гіпогонадизму, які й включають деформації скелета, збої обміну речовин.
Підтвердження розладу починається з огляду. Лікар збирає докладний анамнез. Наявність у родичів будь-яких хромосомних аномалій свідчить про формування генетичного дефекту. Діагноз ставиться на основі специфічної клінічної картинисиндрому Прадера-Віллі, а також результатів каріотипування пацієнта Для виявлення супутніх патологій та складання подальшої терапії проводяться стандартні аналізи крові та УЗД, що дозволяють зробити фото внутрішніх органів, оцінити їх структуру та розміри.
![]()
Специфічних методів боротьби із патологією не розроблено. Ця проблема пов'язана з генетичною основою захворювання. Лікування синдрому Прадера-Віллі носить симптоматичний характер і спрямоване як на коригування порушень, що сформувалися, так і на профілактику розвитку ускладнень.
У дитинстві пацієнтам часто потрібне зондове харчування, а також проведення штучної вентиляціїлегень за наявності респіраторної недостатності. При виявленні гіпотонусу використовуються масажні технікита фізіотерапія, що дозволяють підтримати опорно-руховий апарат.
У ході дорослішання дітям призначаються гормональні засоби. Застосовуються препарати соматотропіну, тестостерону та естрогену залежно від статі пацієнта. Лікувальні заходиспрямовані також на своєчасну та інтенсивну соціалізацію дітей. Вона передбачає спілкування з психіатром, відвідування логопеда та дефектолога. Тривалість терапії індивідуальна і від ступеня вираженості змін. У ряді випадків проводиться оперативне втручаннякорекції дефектів опорно-рухового апарату. Хірургічні техніки використовуються при виявленні вроджених вад серця. У реабілітаційний періодзастосовуються різні медикаментозні засоби. Призначаються адреноблокатори, такі як Енап, ноотропи, до яких відноситься Пірацетам і седативні речовини, наприклад, Персен.
У дитинстві лікарі рекомендують приділяти особливу увагу забезпеченню повноцінного харчування. Це необхідно для адекватного зростання малюка та розвитку внутрішніх органів. Для цього він встановлюються графіки годівлі, і навіть використовуються спеціальні пристрої, що полегшують процес ссання у дітей зі зниженими рефлексами Характерною рисоюсиндрому Віллі-Прадера є тимчасовість проблем із харчуванням, однак у ряді випадків дитині потрібна установка назогастрального зонда. При цьому важливою умовою адекватного годування є контроль калорійності раціону, особливо в період активного зростання. Консультація дієтолога допоможе правильно скласти щоденне менюдитини, що необхідне профілактики ожиріння. Широко використовуються і вітамінно-мінеральні добавки, що забезпечують правильний розвитокопорно-рухового апарату.
Для зниження інтенсивності когнітивних порушень рекомендуються спеціальні стимулюючі техніки. Вони спрямовані на вдосконалення дрібної моторикита мовних навичок. Важливим етапомлікування є і вправи, які дозволяють зміцнювати м'язи та сприяють зменшенню прояву гіпотонії.
У міру дорослішання пацієнта потрібно привчання дитини до контролю власного харчування. Це пов'язано із постійним почуттям голоду на тлі ендокринних порушень. Необхідно дотримання чіткого режиму їди, а також обмеження розмірів порцій.
Хворі потребують сторонньої допомогита підтримки у багатьох аспектах життя. Сім'я має сприяти взаємодії дитини із соціумом, а також заохочувати регулярні фізичні навантаження. Багато пацієнтів потребують спілкування з психотерапевтом для корекції когнітивних порушень, агресії та інших неврологічних дефектів.
Результат захворювання залежить від тяжкості його клінічних проявів, а також своєчасності надання медичної допомоги. За відсутності вад серця, порушення функції нирок та легень пацієнти доживають до похилого віку за умови адекватного лікування.
Специфічних методів профілактики недуги не розроблено. Попередження формування патології засноване на генетичному аналізі каріотипу майбутніх батьків та правильному плануваннівагітності.
Синдром Прадера-Віллі - генетичне захворювання, що виникає внаслідок появи мутації у 15 хромосомі батька.
Різні варіанти зміни генетичного матеріалу батька, що призводять до розвитку захворювання (М – мати, О – батько)
До аномалій відносяться:
Примітка. Крім синдрому Прадера-Віллі, існує подібний синдром Ангельмана. Відмінність цієї хвороби у тому, що описані порушення відбуваються над батьківській, а материнській хромосомі.
Патогенез цього захворювання до кінця не вивчений, проте при аналізі клінічної картини було помічено, що більшість симптомів виникають внаслідок порушення функції гіпоталамуса.
Перші симптоми синдрому Прадера-Віллі з'являються ще до пологів. Першим сигналом стає наявність багатоводдя. Діагноз ставиться з допомогою УЗ дослідження. Наслідок великої кількості навколоплідних вод – неправильне розташування дитини в матці.

Важливо! Необхідно пам'ятати, що, окрім синдрому Прадера-Віллі, ще ряд захворювань можуть викликати багатоводдя, наприклад, інфекційні, резус-конфлікт, вади розвитку та інше. Цей симптом не є високоспецифічним.
У неонатальному періоді дитина буде відрізнятися млявістю, слабким криком, поганим смоктанням. Усе це пов'язано із гіпотонією м'язів.

До початку статевого дозрівання синдром Прадера-Віллі у дітей проявляється затримкою розумового та фізичного розвитку. Дитина зазнає труднощів у навчанні, швидко втомлюється.

У віці 10 – 15 років клінічними симптомами стають:
Важливо! Більшість людей, які страждають на синдром Прадера-Віллі, мають легку розумову відсталість (близько 40%). У 5% пацієнтів – середній рівень інтелекту. 20% мають прикордонні значення IQ між нормою та затримкою. Глибокою розумовою відсталістю страждають менше 1%.

Причиною масивного ожиріння вважається підвищений рівеньгреліну. Цей гормон синтезується гіпоталамусом та відповідає за почуття голоду. Чим вищий його рівень, тим сильніше хочеться їсти. У людей із синдромом Прадера-Віллі кількість греліну в кілька разів збільшена, що робить їх схильними до переїдання.
Примітка. Перші ознаки ожиріння з'являються ще у віці близько двох років.
Через порушення роботи гіпоталамуса не відбувається достатньої стимуляції гіпофіза, і рівень гормонів росту та статевих дуже низький. Це призводить до безпліддя через недорозвинення репродуктивної системи, і навіть до відсутності ростового поштовху.
Незважаючи на велика кількістьСимптомів, діагноз синдрому Прадера-Віллі ставиться досить рідко. За статистикою, близько 2/3 людей, які мають цю генетичну аномалію, залишаються без належного медичного висновку.
Найбільш рання діагностикапроводиться на допологовому етапі. Однак для неї необхідні суворі свідчення, наприклад наявність дітей або близьких родичів з даною хворобою, багатоводдя.

Важливо! Амніоцентез - це інвазивний методдіагностики, що має ряд ускладнень аж до викидня або передчасних пологів. Тому треба ретельно зважити необхідність цього дослідження.
Діагноз синдрому Прадера-Віллі зазвичай встановлюється клінічно вже у віці близько 10-12 років. На той час формується певний габітус (зовнішній вигляд) дитини, і навіть починається затримка зростання та статевого розвитку. Підтверджується клінічний діагнозгенетичним аналізом.
Синдром Прадера-Віллі, як і будь-яке генетичне захворювання, невиліковний. Однак можливе зменшення прояву клінічних симптомівта покращення якості життя пацієнта. При ранній постановці діагнозу починається корекція росту та статевого розвитку за допомогою синтетичних аналогівсоматотропіну та статевих гормонів.

Для боротьби з гіпотонією м'язів проводиться фізіотерапія та масаж. Для корекції проблем із диханням, особливо у нічний час, використовується апарат допоміжної назальної вентиляції легень.
Важливо! Діти мають отримувати психологічну підтримкувід батьків у питаннях адаптації до дитячому колективіта навчанні.
Тривалість життя людей із синдромом Прадера-Віллі залежить від різних факторів. Насамперед - від кількості надмірної ваги. У середньому люди, які страждають на дану хворобу, доживають до 60 і більше років, проте ожиріння може призвести до ранньої загибелі від серцево-судинних захворювань, а також через порушення дихання. Тому люди з цим захворюванням повинні регулярно спостерігатися у лікарів та дотримуватися суворої дієти.

Сенс життя пов'язаний із питанням «Заради чого жити», а не питанням про те, як підтримувати життя. Ставлення людини як...

Гриб - це живий організм, що утворює окреме однойменне царство. Довгий час їх відносили до царства рослин. Але в...

Для любителів «тихого» полювання грибна пора починається раннім літом і триває до пізньої осені. І рідко коли вони...

Олешнікова, В.І. Використання послуг професійних консультантів. - М.: Інфра-М, 1999. - 240 с. 2. Бейч, Е.

Апельсиновий сік. Символічне значення апельсинового соку в сонниках - насолода та спокуса. Досить часто нам...

Різні труднощі, зустрінуті на життєвому шляху, ми найчастіше долаємо. Звичайно, для цього ми додаємо...

Цього року Ваш покровитель Нептун перебуватиме у Вашому сузір'ї і це гарний знак, адже Ви...

1993 кого? 1993 який тварини? — За китайським гороскопом 1873, 1933, 1993 стосувалися років Чорного...

Корпускулярно-хвильовий дуалізм світла означає, що світло одночасно має властивості безперервних...

Роль біології величезна у світі. Хоча вона не входить до пріоритетних предметів, більшість школярів і...
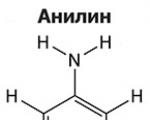
Аміни - органічні похідні аміаку, що містять аміногрупу NH 2 та органічний радикал. У загальному випадку...
Як відповідати на питання частини Друга частина роботи зі суспільствознавства складається з 7 завдань з короткою відповіддю.

Форма ТОРГ-15 складається в тому випадку, коли під час транспортування, переміщення між і всередині складу, при...

Дієтологи кажуть, що для хорошого здоров'я та стрункої фігури, потрібно обов'язково включати перекушування до свого...

Гриб - це живий організм, що утворює окреме однойменне царство. Довгий час їх відносили до царства.

Для любителів «тихого» полювання грибна пора починається раннім літом і триває до пізньої осені. І рідко коли вони...