The most complete interpretation of sleep orange
Orange juice. The symbolic meaning of orange juice in dream books is pleasure and temptation. Quite often we...
Students, graduate students, young scientists who use the knowledge base in their studies and work will be very grateful to you.
Posted on http:// www. allbest. ru/
Ministry of Health of the Republic of Belarus
Belarusian State Medical University
Department of Biology
Hemophilia A, B, C, D as hereditary disease person
Prepared
1st year student
Faculty of Medicine
Groups 103
Budko Yana Andreevna
Minsk, 2015
Introduction
Hemophilia (from ancient Greek b?mb - “blood” and other Greek tsilYab - “love”) is a rare hereditary disease associated with a violation of coagulation (the process of blood clotting). This disease causes bleeding in the joints, muscles and internal organs, both spontaneous and as a result of injury or surgical intervention.
With hemophilia, the risk of death of the patient from hemorrhage in the brain and other vital areas increases sharply. important organs, even with minor injury. Patients with severe form hemophilia becomes disabled due to frequent hemorrhages in the joints (hemarthrosis) and muscle tissue(hematomas). Hemophilia refers to hemorrhagic diathesis caused by a violation of the plasma component of hemostasis (coagulopathy).
1. History
Hemophilia is a disease whose existence has been known for three millennia. The first written mention of it dates back to the 2nd century BC. found in the Talmud Bavli Mesechet Yevamot. She is then mentioned in the 2nd century AD. Rabbi Simon Gamaliel, who describes it as a fatal hemorrhagic disease. In the 12th century, Maimonides and Albucasis noted its familial character. In 1803, John Otto identified the clinical essence of this disease, and in 1820 Nasse described its hereditary transmission. In 1928, Friedrich Hopf gave it the name "haemorraphilie", which in 1839 was shortened to hemophilia. In 1953, M. Biggs divided the hitherto single disease into two variants, A and B.
The most famous person with hemophilia in history was Queen Victoria; Apparently, this mutation occurred in her genotype de novo (for the first time), since there were no cases of hemophilia in her parents' families. Theoretically, this could also happen if Victoria's father was not actually Edward Augustus, Duke of Kent, but some other man (with hemophilia), but there is no historical evidence in favor of this. One of Victoria's sons (Leopold, Duke of Albany), as well as a number of grandchildren and great-grandchildren (born of daughters or granddaughters), including the Russian Tsarevich Alexei Nikolaevich, suffered from hemophilia. For this reason, this disease received the names: “Victorian disease” and “Royal disease”. Also, sometimes in royal families, in order to maintain the title, marriages between close relatives were allowed, which is why the incidence of hemophilia was higher.
2. Genetic aspects diseases
As you know, blood clotting is ensured by 13 factors. Violations of at least one of them lead to various diseases, which includes hemophilia. Depending on which blood clotting factor is impaired, as well as on the mutated gene and mode of inheritance, hemophilia is divided into 4 types.
Hemophilia A occurs due to a recessive mutation in the X chromosome, causing a deficiency of an essential protein in the blood - the so-called factor VIII (antihemophilic globulin). This hemophilia is considered classic; it occurs most often, in 80-85% of patients with hemophilia. Heavy bleeding in case of injuries and operations, they are observed at the level of factor VIII - 5-20%.
Only men are susceptible to hemophilia A, since this disease is associated with the X chromosome, and women in this case are only a transmission link. But, for several months after childbirth, women may also experience prolonged bleeding caused by high level antibodies in the blood to factor VIII. Distinctive feature Hemophilia A is that with this disease, a disruption of the plasma phase of hemostasis occurs in the body. That is, bleeding may not occur at all on the first day after injury, since the first (platelet) and second (vascular) phases function normally. But after a day, suddenly, hemorrhage may begin, which can be extremely difficult to stop, and it can continue for several months, since the blood does not have the ability to clot.
Symptoms: extremely prolonged bleeding various localizations, repeated and delayed bleeding. Typical hemorrhages in hemophilia A include hemorrhages in the muscles and joints, which, without proper treatment, can cause progressive pathological joint and muscle changes (artopathy and atrophy). And this, in turn, leads to human disability. Also, symptoms of hemophilia A may include the occurrence of retroperitoneal, subperiosteal, intramuscular hematomas and pathological bone fractures.
Prevention: none primary prevention hemophilia. In this case, it is important to prevent any types of hemorrhages and bleeding. That is, patients with hemophilia need to avoid intramuscular injections so as not to cause hematomas. All medications should be taken intravenously or orally. Physical activity for patients with hemophilia of any group is strictly prohibited. Children with hereditary hemophilia need to develop intellectual abilities so that you don’t have to do physical labor in the future.
Treatment: the patient is prescribed for life replacement therapy antihemophilic globulin concentrates. If necessary, other hemostatic drugs are also used, but they are only auxiliary elements in the treatment method.
Hemophilia B is a type of hemophilia characterized by a deficiency of clotting factor IX (Christmas factor) in the blood. Just like hemophilia A, B has hereditary causes occurrence. Of the total number of cases of hemophilia, approximately 20% are classified as group B hemophilia.
In medicine, hemophilia B or Christmas disease is usually understood as a hemorrhagic diathesis. This disease was first described back in 1952. Like hemophilia A, it is associated with the X chromosome (a recessive mutation), but, unlike hemophilia A, the structural gene for factor IX is located at the opposite end of the chromosome.
In hemophilia B, the pathology is in the plasma phase of hemostasis. That is, bleeding does not begin immediately after the injury, but after a long time, and continues for a very long time, due to pathology in the function of blood clotting.
Symptoms: joint hemorrhages (hemarthrosis), which are accompanied by pain and a feverish state of the patient. Most often, the elbows, knees and ankle joints. A little less often - the joints of the hands and feet, hip and shoulder. Hemorrhages in the bone tissue, necrosis and bone decalcification. Extensive hematomas that appear suddenly are characteristic of intermuscular hemorrhages. With such hematomas, intense pain occurs caused by blood clots squeezing peripheral nerve trunks or large arteries. This can lead to gangrene or paralysis. Also symptoms of hemophilia B are prolonged and sudden nosebleeds and gum bleeding.
Prevention: designed only to prevent relapses of the disease (similar to hemophilia A). That is, if treatment is properly planned and organized, then patients with hemophilia will not have any symptoms that distinguish them from healthy people.
Treatment: blood transfusions for hemophilia of this group are contraindicated. Dry or frozen donor plasma in this case is more appropriate, since it perfectly preserves the necessary Christmas factor, and in large quantities. Plasma transfusion helps relieve acute joint hemorrhages and minor post-traumatic or postoperative bleeding. When plasma is transfused, factor IX levels increase by almost fifteen percent. But this is not enough to stop larger-scale hemorrhages.
Therefore, in modern medicine, concentrates of the necessary factors are used (in this case - IX), in dosages depending on individual characteristics the patient's body.
Hemophilia C (factor XI deficiency, or Rosenthal syndrome) is characterized by moderate bleeding, occasionally hemarthrosis or hematomas, as well as postoperative bleeding. Clinically, factor XI deficiency is asymptomatic.
In contrast to hemophilia A and B, there is no clear correspondence between bleeding and the level of factor XI, spontaneous bleeding is less pronounced, and hemarthrosis is rare. The diagnosis is often made only when there is bleeding after injury or surgery; Women with factor XI deficiency have heavy and prolonged periods. It has been shown that the number of mutations in the factor XI gene is relatively small. The first such mutation was registered in 1989. To date, thirteen mutations have been identified - 9 missense mutations, 3 splicing mutations and 1 small deletion. Factor XI deficiency is inherited in an autosomal recessive manner and is characteristic of Ashkenazi Jews.
Treatment: transfusion of blood plasma or concentrates containing the missing factor.
Hemophilia D is a factor XII deficiency (Hageman factor deficiency) that causes congenital coagulation disorders in humans - a type of congenital coagulopathy that is usually not accompanied by obvious symptoms. Factor XII deficiency is usually detected in hospital practice when determining blood clotting time before surgery. In women with factor XII deficiency, high risk repeated spontaneous abortions. The type of inheritance is autosomal dominant. The first mutation in the factor XII gene was identified in 1994.
Treatment: similar to the previous one.
hemophilia hereditary disease genetic
3. The situation in the Republic of Belarus
The frequency of hemophilia A in the population (as established by WHO) is 1/100,00, and the percentage of hemophilia type A among all types of this disease is 80-90% (exception: residents of the Teepa Valley in Switzerland). Hemophilia B is 10-15% (population ratio 1:60,000), and type C is 1-2%. As for hemophilia C, its prevalence in the United States is 1:552,000 people. Hemophilia D is the least common. In the animal kingdom, hemophilia occurs in dogs and horses.
The number of such patients in Belarus, according to incomplete estimates, is about 800 people. It should be noted that this figure has increased over the past few years. Thus, in 2007, 463 patients with hemophilia A and 96 patients with hemophilia B were registered.
Unfortunately, there is currently no cure. All measures taken by doctors only help to prolong the lives of those who suffer from this disease.
Literature
1. https://ru.wikipedia.org/wiki/Hemophilia
2. http://www.mednovosti.by
3. http://medbiol.ru/medbiol/blood_dis
4. http://meduniver.com/Medical/gematologia
5. http://nebolet.com/bolezni
6. http://belabg.org
Posted on Allbest.ru
...general information about classical hemophilia and its clinical syndromes. Genetic types hemophilia. A mild form of the disease. Moderately expressed and severe forms diseases. Features and main stages of treatment. The importance of replacement therapy.
abstract, added 03/24/2009
Studying the characteristics of hemophilia, a rare genetic blood disease. Etiology and pathogenesis. Analysis of forms of hemophilia depending on the concentration of antihemophilic factor. Differential diagnosis, replacement therapy and disease prevention.
presentation, added 05/29/2016
Definition of hemophilia as a disease of the group of hemorrhagic diathesis. Genetic basis hereditary coagulopathy - blood clotting disorders. Danger of patient death from cerebral hemorrhage. Treatment and preventive measures.
course work, added 03/11/2011
Rare hereditary diseases, their chronic and progressive course. Concept, types and main causes of hemophilia. Clinical picture, characteristic external symptoms, diagnosis, specialized treatment and prevention of bleeding.
abstract, added 06/05/2016
Classification and differentiation of hereditary diseases. Genetic and chromosomal diseases, diseases with hereditary predisposition. Human genetic maps, treatment and prevention of some hereditary diseases. Description of the main diseases.
presentation, added 11/16/2011
Clinical picture, history of von Willebrand disease - a hereditary blood disease characterized by the occurrence of episodic spontaneous bleeding, which is similar to bleeding in hemophilia. Clotting method of determination (coagulometry).
presentation, added 05/17/2016
Classification of hereditary human diseases. Genetic, mitochondrial and chromosomal diseases. Damage to the hereditary apparatus of the cell. Overall frequency of gene diseases in human populations. Signs of Marfan syndrome and treatment methods for hemophilia.
presentation, added 12/06/2012
Study of blood coagulation factors. Theory of the three-phase process of hemocoagulation. Pathogenetic classification hemorrhagic diseases. Forms of hemophilia, clinical manifestations, frequency of the disease. Methods of treatment and prevention.
abstract, added 09/15/2010
Research on the importance of daily motor activity. Complex physical exercise for knee, ankle, elbow joints for hemophilia. Rehabilitation measures for intramuscular hemorrhages. Eye muscle training and ear massage.
abstract, added 11/03/2014
Hereditary diseases caused by chromosomal and gene mutations. Risk factors for hereditary disease. Prevention and medical genetic counseling. Symptomatic treatment of hereditary diseases. Correction of a genetic defect.
Hemophilia is a hereditary disease in which there is a change in the process of blood clotting and a violation of coagulation. The patient may experience bleeding in different parts his body. This can even happen in the joints.
The disease is also characterized by increased bleeding, or otherwise called blood disease. Hemophilia belongs to the group of hemorrhagic diathesis. Let's take a closer look at the disease.
An anomaly in which the blood clots very poorly due to changes in blood factors and the body's bleeding increases is called hemophilia. This disease belongs to the classification of hemorrhagic diathesis, in which changes in the blood clotting process are considered normal. Cases have been recorded where patients with this disease died very quickly due to sudden bleeding.
The cell nucleus contains chromosomes. They store all the data about a person. Similarity with parents is determined by a special genetic code, which is a member of the chromosome. When gene changes occur, the gene mutates and diseases occur. 23 pairs of chromosomes store all the data about any person. Then this information is passed on hereditarily. Sex chromosomes are the last pair of a number of chromosomes, let's denote them by the letters X and Y. In women, these are XX chromosomes, and in men, XY.

The X chromosome contains a gene that has mutated. It is also responsible for the hereditary transmission of hemophilia. Will the child necessarily suffer from the same hereditary diseases what is his parent? Not at all.
Every child who is born has two genes. They are responsible for one of the characteristics, for example, hair color. One from the mother and the other from the father. There are dominant and recessive genes. That is, dominant (or strong) and secondary (weak). If this happened with two dominant genes, then the strongest one will appear. The same applies to the transmission of recessive traits. If hemophilia is inherited according to only one trait, then the dominant gene will appear as stronger.
The mutated gene, through which the disease is transmitted, is recessive. And it is transmitted only with the X chromosome. The disease will be inherited by a child if he has two recessive genes mutated by this disease with two X chromosomes. When this phenomenon occurs, the baby dies at 4 weeks of pregnancy. After all, it is then that it is formed circulatory system. If only one X chromosome with the trait of hemophilia is transmitted, then the recessive gene will be suppressed by the dominant one, and will not be inherited by the person.

Therefore, women do not suffer from this disease, but are only carriers. The essence of a disease such as hemophilia, the causes of which only a geneticist will accurately reveal, is a blood clotting disorder.
Types of hemophilia have been divided depending on changes in factors affecting clotting.
The severity of hemophilia can be determined by the level of blood clotting:
Hemophilia is congenital disease. Therefore, newly born children have this disease. Signs of hemophilia in newborns include: bleeding from the umbilical cord, various types subcutaneous hematomas. All this is due to the fact that in those suffering from this disease, the vessels are very susceptible to various, even the most minor, damage. When baby teeth grow, they can injure the tongue, lips, and cheeks. Also similar phenomenon may cause bleeding from the oral mucosa. But in newborns who are on breastfeeding, hemophilia rarely manifests itself. Because active thrombokinase contained in breast milk temporarily blocks the development of the disease. Therefore, women who have just given birth are recommended to breastfeed their babies.
Once a child stands up and takes his first steps, post-traumatic bleeding can become very common. This is how hemophilia manifests itself. This disease in a child after a year can manifest itself as frequent nosebleeds, hematomas throughout the body, as well as hemorrhages in large joints. In a child with hemophilia, after suffering from infections (chickenpox, ARVI, rubella, measles, influenza), an exacerbation of hemorrhagic diathesis is possible. As a consequence, vascular permeability is impaired. Then diapedetic hemorrhages can often occur. Also, due to very frequent bleeding, anemia occurs, which occurs in different degrees.

Hemorrhages in hemophilia manifest themselves in different ways. In 70-80%, blood can accumulate in the joints. This phenomenon is called hemarthrosis. Hematomas occur in 10% of cases. Blood is detected in the urine in 14-20%, 8% of patients may have bleeding in the gastrointestinal tract, and 5% of patients are susceptible to hemorrhage in the central nervous system.
Hemarthrosis occurs most often. It is believed that hemophilia manifests itself in this very specific way. This disease is quite insidious. And for the first time such hemorrhages occur in children aged from one to 8 years after bruises and injuries. It can also appear spontaneously. Hemarthrosis is expressed by an increased volume of the joint, the presence pain syndrome. Hyperthermia of the skin also appears. Constant occurrences hemarthrosis in hemophilia is fraught with the occurrence of a chronic inflammatory process in the joint, as well as its deformation and limitation of movements. Such consequences lead to disruption of the dynamics of the musculoskeletal system, and disability occurs in childhood.
Hemophilia is a disease that is manifested by the occurrence of hemorrhages in soft fabrics, that is, into the subcutaneous tissue and muscles, constant bruises that remain on the body, and non-absorbable and intermuscular hematomas. The latter spread because blood does not clot as needed. Therefore, blood penetrates through the tissues. The consequence of the development of such hematomas is severe pain, paralysis, muscle atrophy, or gangrene due to blood accumulations compressing large arteries or nerve trunks.
Hemophilia is characterized by increased bleeding of the body. Moreover, even ordinary intramuscular injection may cause bleeding. This can be dangerous, both for health and for life. For example, bleeding from the pharynx and nasopharynx can lead to obstruction respiratory tract. Then an emergency tracheotomy will be needed. Severe damage to the central nervous system can result from cerebral hemorrhages.
Spontaneously or due to injury to the lumbar region, blood in the urine can be caused. In this case, the process of urination is disrupted. Then the blood coagulates in such a way that blood clots in urine. Hemophilia can often cause kidney diseases that are characterized by severe inflammatory process, which will subsequently affect the functioning of the entire body.
If you take anti-inflammatory and antipyretic medications, gastrointestinal bleeding may occur if the patient has hemophilia. As a result, a stomach ulcer may worsen, duodenum, erosive gastritis, haemorrhoids. Bleeding in hemophilia after injury may not occur immediately, but only after 6-12 hours.
The child is examined by several doctors: a neonatologist, a pediatrician, a geneticist and a hematologist. And only after this is hemophilia diagnosed. If a child has a pathology or complication of the underlying disease, then an examination by pediatric doctors is necessary: a gastroenterologist, an orthopedic traumatologist, an otolaryngologist and a neurologist to monitor how hemophilia develops. What kind of disease this is can be found out from a doctor specializing in blood diseases.
If you are planning to have a child, then future parents who are at risk should consult a geneticist. This should be done before pregnancy has been determined. Genealogical data and molecular genetic diagnostics determine who is the carrier of the defective gene. Hemophilia can be detected through perinatal diagnosis, using a chorionic villus tissue sample or amniocentesis to examine the DNA of the cellular material. Note that the reasons associated with abnormal development of the fetus are not addressed in this case.
To confirm the diagnosis of hemophilia, hemostasis is examined immediately after the baby is born. If hemophilia is present, then the coagulogram parameters will differ from the norm.

This will usually be noticeable by how slowly the blood clots. In hemophilia, as a rule, the procoagulant activity of one of the coagulation factors is reduced.
In case of hemorrhages due to hemophilia, the child is prescribed various studies of organs, joints, general analysis urine, blood.
There is no complete cure for hemophilia. Therefore, the basis of treatment is usually therapy aimed at improving blood clotting. That is, factors VIII and IX are replaced with their concentrates. They affect the process of blood clotting. First, determine what dose of concentrate is needed. This can be determined by the severity of the disease, its form, and also by the type of bleeding in the patient.
Hemophilia can be treated during its manifestation hemorrhagic syndrome and simply carry out prevention. A severe form of the disease requires prophylactic administration of the necessary concentrates. Such therapy should be carried out 2-3 times a week so that secondary joint damage does not develop.
The use of fresh frozen plasma, erythromass, hemostatics, as well as repeated transfusions of drugs is necessary to stop the development of hemorrhagic syndrome. Hemostatic therapy is also necessary when any intervention is required, so that nothing will cause complications that will aggravate hemophilia. We have already found out what kind of disease this is.
Even with the smallest hemorrhages, serious measures must be taken. Because in hemophilia it can become fatal.

If it is a cut, then you can apply hemostatic sponge, a pressure bandage and treat the wound with thrombin. Complete rest is needed even when an uncomplicated hemorrhage occurs. In this case, the patient should be placed in a cool room, and the damaged joint should be fixed with a splint. In the future, his condition should be monitored using diagnostics. Maintaining a certain diet is also important. Food products should be enriched with vitamins A, B, C, D and calcium and phosphorus salts.
If you carry out therapy for a long time, which consists of administering concentrates of the missing factors, the body will begin to produce antibodies. The latter will block the effectiveness of the introduced factors. In such cases, hemostatic therapy becomes ineffective.

Therefore, immunosuppressants and plasmapheresis are additionally prescribed. Patients with hemophilia often receive blood transfusions, and there is a risk of contracting HIV infection, hepatitis B, C, D. You need to be very careful about this event, find out if all checks have been carried out.
Life expectancy does not change if patient's lung the degree of hemophilia, but if not, then the consequences of massive bleeding can affect not only its quality. Therefore, people with hemophilia can live for a short period of time.
Also to preventive measures hemophilia includes consultation in the field of medical genetic research of married couples who have relatives suffering from this disease.
Children with this diagnosis should always have a passport with the specified disease, blood type and Rh factor. Such babies should be observed by a pediatrician, hematologist and other doctors.
In addition, people with this disease must be registered with specialized medical personnel. They need to be constantly monitored in order to monitor the dynamics of hemophilia. Also for selection and use the latest methods treatment of this disease.
Now you know what hemophilia is, the reasons for its occurrence are known to you. We also looked at the symptoms of the disease, routes of transmission, diagnosis, as well as methods of maintenance therapy.
Hemophilia is a hereditary disease associated with a defect in plasma clotting factors, characterized by a disorder of blood clotting. Known this pathology since ancient times: back in the 2nd century BC, cases of boys dying from incessant bleeding occurring after the circumcision procedure were described. The term "hemophilia" was coined in 1828 and comes from the Greek words "haima" - blood and "philia" - tendency, that is, "tendency to bleed."
This disease occurs with a frequency of 1 case per 50,000 newborns, with hemophilia A diagnosed more often: 1 case of the disease per 10,000 people, and hemophilia B less often: 1: 30,000-50,000 male residents. Hemophilia is inherited through a recessive trait associated with the X chromosome. In 70% of cases, hemophilia is characterized severe course, steadily progresses and leads to early disability of the patient. The most famous hemophiliac in Russia is Tsarevich Alexei, the son of Alexandra Feodorovna and Tsar Nicholas II. As you know, the disease passed to the family Russian Emperor from his wife's grandmother, Queen Victoria. Using the example of this family, the transmission of the disease along the genealogical line is often studied.
Family tree of Queen Victoria
Blood coagulation is a set of complex biochemical processes and reactions, the purpose of which is to stop bleeding in the event of damage to the vessel wall. The main role in this process belongs to the so-called blood clotting factors.
Conventionally, the entire process of blood clotting can be divided into 3 stages:
There are only 13 plasma blood clotting factors. When the level of at least one of them decreases in the body, normal blood clotting becomes impossible.
As mentioned above, hemophilia is hereditary pathology. It is caused by a mutation in a gene that controls the synthesis of a particular blood clotting factor.
As a result of factor deficiency, the formation of a normal blood clot does not occur, that is, the bleeding that has developed does not stop in due time.
 Depending on which coagulation factor is missing in the body, there are 3 types of hemophilia.
Depending on which coagulation factor is missing in the body, there are 3 types of hemophilia. Currently, there are 3 forms of hemophilia:
The leading clinical sign of this disease is increased bleeding from the very first days of the baby’s life. This manifests itself with all kinds of bruises, cuts and other interventions. Deep hemorrhages and hematomas appear, and prolonged bleeding occurs when teeth erupt and fall out.
In older age of the patient, the main symptom also becomes spontaneous or occurring after injuries. heavy bleeding or hemarthrosis: hemorrhages in large joints. The joint with a hematoma is enlarged, swollen, and sharply painful. Repeated hemarthrosis causes inflammatory changes in the joint of a secondary nature, which subsequently lead to contractures (limitation of the range of passive movements) and ankylosis (complete immobilization of the joint). As a rule, changes affect only large (knee, elbow, ankle) joints, and small ones (for example, hand joints) are affected much less frequently. As the disease progresses, the number of affected joints also increases: pathological process up to 12 joints may be involved at the same time. Often this becomes the cause of early – even at the age of 15-20 years – disability of the patient.
Hematomas can occur not only in the joint area. Cases of subfascial, intermuscular and retroperitoneal hematomas are common. The volume of blood that makes up hematomas can be relatively small - 0.5 liters, but can reach impressive figures - even up to 3 liters.
If the hematoma is so large that it compresses a nerve or blood vessel, the patient experiences intense pain, signs of ischemia of one or another organ, in varying degrees limitation or complete loss of voluntary movements: paresis or paralysis.
IN severe cases disease, there is a threat of life-threatening gastrointestinal and/or renal bleeding. The first are manifested by vomiting containing blood (the so-called “vomiting coffee grounds") and black liquid feces. If there is renal bleeding, the patient will notice the red color of the urine.
As mentioned above, women extremely rarely suffer from hemophilia. The nature of its course also depends on the degree of deficiency of the missing factor; clinical manifestations are standard. The only thing is that women with hemophilia, as well as women who are carriers of a pathological gene, have a risk of developing massive postpartum hemorrhage quite high.
The severity of hemophilia directly depends on the degree of reduction in the level of clotting factor in the blood. When it decreases by less than 50%, there are no clinical signs of the disease. With a small decrease (within 20-50% relative to normal values) – bleeding develops after a serious traumatic injury or against the background of surgical intervention. In individuals who carry the hemophilia gene, the level of factor VIII or IX is also slightly reduced. When the factor concentration in plasma decreases to 5-20% of normal values, bleeding occurs against the background of moderate injuries. If the level of the factor is very low and ranges from 1-5% of the norm (this is a severe form of hemophilia), the patient experiences spontaneous bleeding into the joints and soft tissues. Finally, complete absence clotting factor is manifested by massive spontaneous bleeding and frequent hemarthrosis.
The disease is diagnosed based on the patient’s complaints, medical history (bleeding, hemarthrosis, appearing with early childhood; perhaps one of the male relatives suffered from hemophilia), typical clinical signs. When examining a patient with hemophilia, the doctor will pay attention to large joints that are deformed and deformed, with a limited range of motion, and during exacerbation they are also painful. The muscles around the joints are atrophied, the limbs are thinned. There are multiple bruises (hematomas) and pinpoint hemorrhages on the patient’s body.
If hemophilia is suspected, the patient undergoes the following laboratory tests:
Differential diagnosis of hemophilia should be carried out with the following diseases:
 The main direction of treatment for hemophilia is replacement therapy, which involves regular injection of the missing clotting factor into the bloodstream.
The main direction of treatment for hemophilia is replacement therapy, which involves regular injection of the missing clotting factor into the bloodstream. Therapy of the disease has the following goals:
For hemophilia, replacement therapy for the missing plasma clotting factor is carried out. The dose of the drug is determined individually depending on the initial level of the factor in the blood and the patient’s body weight. Since the half-life of the drug is 8-12 hours, it should be administered 2-3 times a day.
Surgical interventions for persons suffering from hemophilia are carried out strictly according to indications against the background of mandatory replacement therapy.
Children suffering from hemophilia should always carry with them a “hemophiliac passport”, which indicates the type of disease, blood type and Rh factor of the patient, as well as the principles of care for him. emergency care. Parents of a child with hemophilia are strongly advised to have a supply of deficiency factor medication.
Patients should be under the dynamic supervision of a hematologist, traumatologist, orthopedist, and also from time to time take control tests blood: general and biochemical.
This pathology is highly treatable. When prescribed in a timely manner, the quality of life of patients improves significantly. In the absence of treatment, it quickly leads to permanent disability of the patient, and some of its complications can even cause his death.
Many diseases that we may encounter in our lives are passed on to us along with genes from our parents and more distant relatives. In addition, a certain predisposition to certain diseases can also be hereditary. As practice shows, such illnesses are quite rare, but they pose a serious threat to human health and even life. One of the most dangerous diseases of this type is considered to be hemophilia, which affects only boys. How can this disease be identified? What is the diagnosis of hemophilia? We will also talk about how hemophilia is inherited and how it is transmitted.
In general, hemophilia can be characterized as a tendency of the human body to prolonged hemorrhages, as well as bleeding. In this case, the patient’s bleeding develops sharply without any reason, and they can only be stopped by using special drugs.
How is hemophilia diagnosed? Diagnosis of the disease
To make a diagnosis of hemophilia, it is necessary to conduct a series of laboratory tests that can accurately indicate the corresponding changes in the body. In addition, the doctor takes a family history, which helps determine the likelihood of developing the disease, and carefully examines clinical picture diseases.
Concerning laboratory methods research, they include the determination of several indicators:
Recording the amount of coagulation factors inside the blood;
- determination of the time interval of coagulation;
- level of fibrinogen in the blood;
- prothrombin index;
- thrombin time;
- international normalized ratio;
- mixed – APTT;
- activated partial thromboplastin period.
If a person has hemophilia, there is an above-average increase normal level indicators such as: blood clotting time, aPTT, thrombin time and international normalized ratio. At the same time, the level of the prothrombin index decreases, but the values of the mixed APTT, as well as the level of fibrinogen, remain within normal limits.
The main indicator of hemophilia A or B is considered to be a decrease in the concentration or adequate activity of coagulation factors in the blood. A decrease in factor VII occurs in hemophilia type A, and a decrease in factor IX occurs in type B disease.
The development of hemophilia can be suspected literally immediately after birth. If the baby does not stop bleeding for a long time after cutting the umbilical cord, and there are also hematomas on the surface of the head, buttocks and perineum, it is extremely important to conduct a test for hemophilia. To date, there is no way to predict such a disease at the prenatal stage. All diagnostic methods in the prenatal period are complex, have contraindications and are often not informative enough. That is why it is extremely important role It is important to collect a family history at the stage of planning a child, as well as conduct a consultation with a qualified geneticist.
How is hemophilia transmitted?
The main feature of hemophilia, as a hereditary disease, is that women are considered carriers of this pathology, and only men suffer from it. What explains this?
The hemophilia gene is recessive and is present on the so-called X chromosome. Its inheritance is linked to sex; therefore, in order for it to manifest itself, the presence of a pair of chromosomes with this mutation is necessary. As is known, women have a pair of sex X chromosomes, and men have a combination of X and Y chromosomes. Accordingly, for hemophilia to develop in a woman, she needs two mutated X chromosomes to come together, but this is completely impossible.
If a girl with such a genetic makeup becomes pregnant, a miscarriage occurs already in the fourth week after conception. Pregnancy is terminated after the formation of the fetus's own blood begins; the mother's body simply gets rid of the child as non-viable. That is why girls with only one mutated X chromosome can be born. In this case, the disease does not make itself felt, since the second X chromosome is dominant and suppresses the recessive one, causing the problem with blood. Thus, women can only be carriers of hemophilia.
Males have one X chromosome, and the Y chromosome cannot contain the gene for this disease. Accordingly, if the X chromosome is mutated, then the Y chromosome does not have a second dominant gene that can suppress the pathological recessive one. That is why the disease manifests itself in boys and they are diagnosed with hemophilia.
Unfortunately, today this disease cannot be cured. Doctors can only monitor it, prevent complications if possible and provide supportive treatment. For this purpose, patients are given a special clotting factor that they do not have in their body. This factor is now obtained from donor or animal blood. The right approach therapy can make the life expectancy of a hemophilia patient the same as that of a healthy person. However, constant use donor material significantly increases the likelihood of patients developing AIDS or hepatitis.
At the stage of planning a child, all couples are strongly recommended to visit a geneticist. The specialist will help identify the possible likelihood of having children with hereditary diseases and give the spouses certain recommendations.
In our article today:
Most hereditary human diseases are caused by changes in genes, mutations. This means that diseases must be inherited in the same way as any other characteristics of the body: they can be dominant or recessive, determined by one gene or several.Let us remember that some characteristics of many organisms turn out to be “sex-linked”; their genes are located on the sex chromosomes. But humans also have an X chromosome, which means there must be sex-linked diseases. The most famous of these diseases is hemophilia, or blood inability to clot. In people suffering from this disease, the blood is unable to clot, so even a minor scratch can lead to serious bleeding and even death. , as befits a hereditary disease, it is always inherited. But the patterns of such inheritance are very interesting. First of all, only men suffer from hemophilia; cases of the disease among women are extremely rare. But it is transmitted exclusively through the maternal line. How is this possible? Here it is necessary to remember how blood clots. Blood clotting is very difficult process, it includes several consecutive chemical reactions, each of which involves its own specific enzyme protein (they are called blood clotting factors). Without at least one of them, blood clotting becomes impossible: the person develops hemophilia.
The most common is hemophilia A, or royal hemophilia, in which a person lacks the so-called factor VIII (that is, a protein that is involved in the eighth blood clotting reaction). Less common is hemophilia B, in which case there is a deficiency of factor IX. But since factor VIII is a protein, it means there is a gene that is “responsible” for it. In humans, such a gene is located on the sex chromosome X, while the normal gene (it is designated by the sign dominates over the mutant hemophilia gene (it is usually designated as “0”)). Now it is clear why mostly men suffer from hemophilia; they have only one X chromosome, This means that the recessive gene 0, which is located in it, can manifest itself. And the disease is transmitted through the maternal line because the boy receives his only X chromosome from his mother (from his father he gets the Y chromosome, which does not contain this gene at all). Woman can be a carrier of hemophilia (this means that one of her two X chromosomes contains gene 0), but at the same time be completely healthy (the second X chromosome contains a dominant gene that suppresses the action of the hemophilia gene). if her father has hemophilia and her mother is a carrier. In this case, gene 0 can appear on both X chromosomes. But this happens extremely rarely. Moreover, even if a woman is a carrier of hemophilia, it is not at all necessary that her sons will inherit this terrible disease. After all, she can transmit to them both the “sick” and “healthy” X chromosome with equal probability. Likewise, her daughters will not necessarily be carriers of the disease.
Why is hemophilia sometimes called the royal or royal disease? The fact is that many descendants of Queen Victoria of England suffered from hemophilia on their mother’s side. Tsarevich Alexei, who was the son of Nicholas II, was sick with hemophilia. His mother, the queen Alexandra Fedorovna, was a carrier of hemophilia and inherited it from her mother Alice. And she, in turn, received hemophilia “inherited” from Tsarevich Alexei’s great-grandmother, Queen Victoria. Many princes in the royal families of Europe, descendants of Queen Victoria (as scientists and historians suggest, the gene mutation came from her) received this gene and were sick with hemophilia, while the princesses were carriers of hemophilia, but were themselves absolutely healthy. Can hemophilia be cured? Unfortunately, even modern medicine is powerless against hemophilia. The only way to help patients is to regularly administer ready-made factor VIII protein, obtained from human donors or animals. However, it is quite possible that genetic disease hemophilia will very soon be treated using genetic methods. Scientists have already learned to treat hemophilia in laboratory mice (these rodents also suffer from royal disease) by artificially correcting the “damaged” gene.
Hemophilia is far from the only hereditary human disease that is sex-linked. In addition to both types of hemophilia, chromosome X is “responsible” for some of our other ailments, the most famous of which is color blindness. Colorblind people are people who are unable to distinguish colors: they either generally see the world in black and white, or they confuse different colors together. Color blindness gene, like the hemophilia gene, is located on chromosome X, so color blindness is 20 times more common among men than among women.

Orange juice. The symbolic meaning of orange juice in dream books is pleasure and temptation. Quite often we...

We most often overcome all kinds of difficulties encountered along the path of life. Of course, for this we make...

This year your patron Neptune will be in your constellation and this is a good sign, because you will feel...

1993 who? 1993 is the year of which animal? — According to the Chinese horoscope, 1873, 1933, 1993 belonged to the years of the Black Water...

Wave-particle duality of light means that light simultaneously has the properties of continuous electromagnetic...

The role of biology is enormous in our world. Although it is not a priority subject, most students and parents...
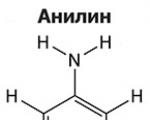
Amines are organic derivatives of ammonia containing an NH 2 amino group and an organic radical. In general...
How to answer questions in Part BThe second part of the social studies work consists of 7 tasks with a short answer....

Form TORG-15 is drawn up in the case when during transportation, movement between and within the warehouse, when...

Nutritionists say that for good health and a slim figure, you must include snacks in your...

Delicious pickled carrots for the winter can be prepared in a variety of containers, it can be a wooden...

When I have a few minutes left to prepare breakfast, the simplest and fastest recipes are used....

An elegant table, a decorated Christmas tree, tangerine spirit spilled throughout all the rooms, soon the most magical holiday -...

Each of us has repeatedly faced financial difficulties and difficult periods in life when Fortune...

We most often overcome all kinds of difficulties encountered along the path of life. Of course, for this we...

This year your patron Neptune will be in your constellation and this is a good sign, because you will...