Visão filosófica do problema
O sentido da vida está relacionado com a questão “Para que viver”, e não com a questão de como manter a vida. A atitude de uma pessoa é...
Esta patologia é caracterizada por desvios na estrutura e funcionamento dos órgãos genitais. A origem da doença ainda não foi estabelecida, mas os médicos acreditam que a síndrome se desenvolve devido à produção excessiva de andrógenos pelo córtex adrenal. A doença pode ser causada por vários tumores ou hiperplasia congênita da glândula.
A hiperplasia adrenal congênita é o tipo mais comum de patologia verulizante. A síndrome adrenogenital é uma doença conhecida na medicina mundial como síndrome de Apère-Gamay. Seu desenvolvimento está associado ao aumento da produção de andrógenos e à diminuição acentuada dos níveis de cortisol e aldosterona, causada por disfunção congênita do córtex adrenal.
As consequências do desvio podem ser graves para o recém-nascido, uma vez que o córtex adrenal é responsável pela produção de uma grande quantidade de hormônios que regulam o funcionamento da maioria dos sistemas do corpo. Como resultado da patologia no corpo da criança (isso pode ser observado tanto em meninos quanto em meninas), há muito hormônios masculinos e muito poucas mulheres.
Cada forma da doença está associada a doenças genéticas: via de regra, as anomalias são hereditárias e passam de ambos os pais para o filho. Mais raros são os casos em que o tipo de herança da síndrome adrenogenital é esporádico - ocorre repentinamente durante a formação de um óvulo ou espermatozoide. A herança da síndrome adrenogenital ocorre de forma autossômica recessiva (quando ambos os pais são portadores do gene patológico). Às vezes, a doença afeta crianças em famílias saudáveis.
A síndrome adrenogenital (AGS) é caracterizada pelos seguintes padrões que afetam a probabilidade de afetar uma criança:
A doença androgenética é convencionalmente dividida em três tipos - viril simples, perdedora de sal e pós-púbere (não clássica). As variedades apresentam grandes diferenças, por isso cada paciente requer um diagnóstico detalhado. Como se manifestam as formas da síndrome adrenogenital:
A disfunção adrenal congênita é explicada apenas pela manifestação de uma doença hereditária, sendo impossível adquirir ou se infectar com tal patologia durante a vida. Via de regra, a síndrome se manifesta em recém-nascidos, mas a AGS raramente é diagnosticada em jovens com menos de 35 anos. Ao mesmo tempo, factores como a toma de medicamentos potentes, o aumento da radiação de fundo, os efeitos secundários da contraceptivos hormonais.
Qualquer que seja o estímulo para o desenvolvimento da doença, as causas da síndrome adrenogenital são hereditárias. A previsão é mais ou menos assim:

AGS não é uma doença mortal, mas alguns de seus sintomas causam sério desconforto psicológico a uma pessoa e muitas vezes levam a colapso nervoso. Ao diagnosticar uma patologia em um recém-nascido, os pais têm tempo e oportunidade para ajudar a criança na adaptação social e, se a doença for detectada na idade escolar ou mais tarde, a situação pode ficar fora de controle.
A presença de AGS só pode ser determinada após testes moleculares. análise genética. Os sintomas da síndrome adrenogenital que indicam a necessidade de diagnóstico são:
A doença pode ser detectada precocemente no recém-nascido, o que está associado à triagem neonatal no 4º dia após o nascimento da criança. Durante o exame, uma gota de sangue do calcanhar do bebê é aplicada em uma tira-teste: se a reação for positiva, a criança é transferida para um ambulatório de endocrinologia e é feito o re-diagnóstico. Assim que o diagnóstico for confirmado, o tratamento para AGS começa. Se a síndrome adrenogenital em recém-nascidos for detectada precocemente, a terapia é fácil; nos casos de detecção tardia da patologia adrenogenética, a complexidade do tratamento aumenta.

A doença geralmente se desenvolve em crianças do sexo masculino a partir dos dois ou três anos de idade. Ocorre um aumento do desenvolvimento físico: os órgãos genitais aumentam, ocorre um crescimento ativo do cabelo e as ereções começam a aparecer. Nesse caso, os testículos ficam para trás no crescimento e, posteriormente, param de se desenvolver completamente. Assim como nas meninas, a síndrome adrenogenital nos meninos é caracterizada por um crescimento ativo, mas não dura muito e no final a pessoa ainda permanece baixa e atarracada.
A patologia em meninas é frequentemente expressa imediatamente no nascimento, na forma viril. O falso hermafroditismo feminino, característico da AGS, é caracterizado por um aumento do tamanho do clitóris, enquanto a abertura uretra está localizado diretamente abaixo de sua base. Os lábios, neste caso, lembram o formato de um escroto masculino dividido (o seio urogenital não é dividido em vagina e uretra, mas para de se desenvolver e se abre sob o clitóris em forma de pênis).
Não é incomum que a síndrome adrenogenital em meninas seja tão pronunciada que, no nascimento de um bebê, seja difícil determinar imediatamente seu sexo. Durante o período de 3 a 6 anos, a criança deixa crescer ativamente pelos nas pernas, na região pubiana e nas costas, e a aparência da menina se torna muito semelhante a um menino. As crianças com AHS crescem muito mais rapidamente do que os seus pares saudáveis, mas o seu desenvolvimento sexual rapidamente pára completamente. Ao mesmo tempo, as glândulas mamárias permanecem pequenas e a menstruação está completamente ausente ou aparece de forma irregular devido ao fato de os ovários subdesenvolvidos não conseguirem desempenhar plenamente suas funções.
A doença pode ser identificada usando pesquisa moderna níveis hormonais e exame visual. O médico leva em consideração dados anamnésicos e fenotípicos, por exemplo, crescimento de pêlos em locais atípicos para uma mulher, desenvolvimento das glândulas mamárias, tipo de corpo masculino, aparência geral/saúde da pele, etc. AHS se desenvolve devido a uma deficiência de 17 -alfa-hidroxilase, para que o sangue do paciente possa monitorar o nível dos hormônios DHEA-S e DHEA, que são precursores da testosterona.
O diagnóstico também inclui um teste de urina para determinar o nível de 17-KS. Um exame bioquímico de sangue permite determinar o nível dos hormônios 17-OHP e DHEA-S no corpo do paciente. Além disso, o diagnóstico abrangente envolve o estudo dos sintomas do hiperandrogenismo e de outros distúrbios do sistema endócrino. Nesse caso, os indicadores são verificados duas vezes - antes e depois do teste com glicocorticosteróides. Se durante a análise o nível hormonal for reduzido para 75% ou mais, isso indica a produção de andrógenos exclusivamente pelo córtex adrenal.
Além dos exames hormonais, o diagnóstico da síndrome adrenogenital inclui a ultrassonografia dos ovários, na qual o médico determina a anovulação (pode ser detectada se forem observados folículos de diferentes níveis de maturidade, não ultrapassando os volumes pré-ovulatórios). Nesses casos, os ovários estão aumentados, mas o volume do estroma é normal e não há folículos sob a cápsula do órgão. Somente após um exame detalhado e confirmação do diagnóstico é iniciado o tratamento da síndrome adrenogenital.

O ABC não é uma patologia fatal com resultado letal, portanto a probabilidade de desenvolver alterações irreversíveis no corpo do paciente é extremamente baixa. No entanto tratamento moderno a síndrome adrenogenital não pode se orgulhar de sua eficácia e eficiência. Pacientes com esse diagnóstico são obrigados a tomar medicamentos hormonais pelo resto da vida para compensar a deficiência dos hormônios glicocorticosteróides e para combater o sentimento de inferioridade.
As perspectivas para tal terapia permanecem inexploradas, mas há evidências de que há uma alta probabilidade de desenvolver patologias concomitantes de AGS no coração, ossos, vasos sanguíneos, trato gastrointestinal e câncer. Isso explica a necessidade de pessoas com disfunção do córtex adrenal se submeterem a exames regulares - radiografias ósseas, eletrocardiograma, ultrassonografia do peritônio, etc.
sovets.net
A síndrome adrenogenital é um grupo de doenças hereditárias e adquiridas cujo principal sintoma é uma mutação em um gene específico. Esses distúrbios se manifestam na secreção excessiva de hormônios do córtex adrenal.
Voltar ao conteúdo
Tanto meninos quanto meninas estão predispostos a esta doença, e igualmente. A síndrome adrenogenital em recém-nascidos é bastante comum. A probabilidade de ocorrência de um defeito enzimático é de cerca de 1:14.000. O diagnóstico tardio da síndrome adrenogenital e a terapia inadequada podem levar a consequências bastante graves:
Voltar ao conteúdo

A síndrome adrenogenital ocorre em três formas:
Aqueles. cada forma tem seus próprios sintomas de síndrome adrenogenital.
Voltar ao conteúdo
A síndrome adrenogenital em recém-nascidos ocorre por vários motivos. Cada um deles tem seu próprio características distintas e depende da forma da doença.
O mais comum é a deficiência de enzimas envolvidas na síntese dos hormônios adrenais - 21-hidroxilase, menos comumente 11-hidroxilase e 3-beta-ol-desidrogenase e outros. Como resultado, são sintetizadas quantidades insuficientes de cortisol e aldosterona. Estas substâncias pertencem aos hormônios esteróides do córtex adrenal. O cortisol é responsável por proteger o corpo contra vários tipos de infecções e estresse. A aldosterona é responsável por nível normal pressão arterial e função renal. A falta desses hormônios leva ao aumento da secreção do hormônio adrenocorticotrófico, que promove hiperplasia do córtex adrenal e o desenvolvimento desta doença.
Voltar ao conteúdo
Porque Esta doença é de natureza congênita, causada por mutação de certos genes, então a única método preventivo para a síndrome adrenogenital, podem ser convocadas consultas médicas de genética.
Voltar ao conteúdo
A síndrome adrenogenital em crianças pode ser detectada numa fase inicial da doença. Isso se deve ao fato de todos os bebês nascidos na maternidade passarem pela triagem neonatal no quarto dia - uma gota de sangue do calcanhar do recém-nascido é aplicada em uma tira teste. No reação positiva a criança é encaminhada ao ambulatório de endocrinologia para reexame. Uma vez confirmado o diagnóstico, o tratamento adequado é imediatamente prescrito.
O diagnóstico da síndrome adrenogenital é bastante simples. Porém, na prática, segundo dados estatísticos, esta patologia é detectada nas fases mais avançadas da doença.
Atualmente, o diagnóstico desta doença inclui as seguintes etapas:
Voltar ao conteúdo
O tratamento da síndrome adrenogenital é realizado de duas maneiras - medicamentosa e cirúrgica.
Método de medicação depende da forma da doença. Assim, na forma viril, consiste em repor a falta dos hormônios glicocorticosteróides. A dose do medicamento (prednisolona) é estritamente individual e determinada pelos seguintes parâmetros - idade do paciente, grau de virilização. Normalmente, a quantidade resultante do medicamento (4 - 10 mg) é tomada em três doses durante o dia. Isso permite suprimir o excesso de secreção de andrógenos sem quaisquer efeitos colaterais.
O tratamento da síndrome adrenogenital na forma perdedora de sal é realizado de forma semelhante à insuficiência adrenal aguda. Nesse caso, utiliza-se a administração gota a gota de soluções isotônicas de cloreto de sódio e glicose. A administração parenteral de hidrocortisona também é utilizada. A quantidade deste medicamento é de cerca de 10 a 15 mg/kg por dia, que é distribuída uniformemente. À medida que a condição se estabiliza, as injeções de hidrocortisona são substituídas por comprimidos. Às vezes, se necessário, pode ser adicionado um mineralocorticóide.
A intervenção cirúrgica é necessária em caso de diagnóstico tardio da doença, se os sintomas da síndrome adrenogenital não forem pronunciados. Em seguida, as meninas de 4 a 6 anos são submetidas à correção da genitália externa.
Voltar ao conteúdo
Nas formas viris e perdedoras de sal, o diagnóstico oportuno, o tratamento adequado e a possível intervenção cirúrgica proporcionam um prognóstico favorável.
Pacientes com síndrome adrenogenital ao longo da vida devem ser acompanhados por endocrinologista, ginecologista ou urologista. Porém, com tratamento adequado, eles continuam aptos a trabalhar.
Voltar ao conteúdo
equipehelp.ru
A síndrome adrenogenital é causada por um defeito enzimático congênito herdado recessivamente.
.Maioria razão comum- defeito da enzima 21-hidroxalase. Devido a um defeito enzimático, nem os glicocorticóides nem os mineralocorticóides são produzidos com frequência.
Violação da síntese de cortisol como resultado da fermentopatia → nível aumentado virilização androgênica
95% de todos os pacientes apresentam defeito da 21-hidroxilase, dos quais 75% apresentam perda de sal devido à deficiência de aldosterona
Meninas recém-nascidas: virilização da genitália externa.
Meninos recém-nascidos: aumento da pigmentaçãoórgãos genitais.
Síndrome de perda de sal durante manifestação primária devido a infecção, trauma, cirurgia.
Principais sintomas da síndrome de perda de sal:
Determinação das concentrações hormonais no sangue, saliva e urina diária.
Tratamento de emergência da crise de perda de sal.
Reembolso de volume:
Esteróides:
Correção de acidose: Bicarbonato de Na em dosagem usual para EB
Terapia de longo prazo de AGS com perda de sais.
Esteróides:
Os bebês recebem 1-3 g adicionais de NaCl por dia (distribuídos uniformemente em todas as mamadas). O leite materno contém muito pouco sódio!
A terapia é realizada ao longo da vida. Atestado de patologia em caso de emergência!
Para estresse, cirurgia, infecções, etc. um aumento de 4 vezes na dose de hidrocortisona:
A prevenção pré-natal da virilização em meninas com AGS é possível através da administração de dexametasona às mães. No entanto, como os efeitos colaterais a longo prazo dessa profilaxia ainda não são conhecidos, esta técnica ainda é considerada experimental.
Administração de gotejamento solução isotônica cloreto de sódio para estabilizar a circulação sanguínea e repor os sais.
Hidrocortisona imediatamente em altas doses, por ex. administração intravenosa solução diluída de hidrocortisona → perigo de tromboflebite, dor.
Se houver perda de sais, adicionalmente mineralocorticóides, como fludrocortisona, em comprimidos.
Monitore o tratamento avaliando o crescimento, determinando a idade óssea e verificando regularmente as concentrações hormonais na saliva, urina e/ou sangue.
Cirurgias corretivas nos órgãos genitais em casos de virilização grave em meninas para garantir o funcionamento normal dos órgãos genitais e a identificação do género.
Forneça à criança e sua família informações detalhadas sobre o problema da insuficiência adrenal. Dê a cada criança com síndrome adrenogenital um documento que ela deve levar sempre consigo (perigo insuficiência aguda córtex adrenal durante operações, infecções, lesões).
Monitoramento, medição especialmente cuidadosa da pressão arterial.
Controle do volume de líquido recebido e liberado, em alguns casos - instalação de cateter permanente.
Cumprimento cuidadoso das regras de higiene.
No exaustão severa- prevenção de escaras e contraturas.
Dependendo da idade e condição geral: informar a criança sobre a doença.
www.sweli.ru
A síndrome adrenogenital é uma patologia muito específica, mas pouco conhecida. De certa forma, pode servir como uma triste ilustração do conto de fadas sobre o czar Saltan, de Alexander Sergeevich Pushkin, mas isso não torna as coisas mais fáceis para os próprios pacientes. Lembra-se das famosas falas, cujo significado a maioria das pessoas mal pensa: “a rainha deu à luz um filho ou uma filha naquela noite”? Tem certeza de que esta é exclusivamente uma metáfora do autor? Nada como isso! Tais “sintomas” ocorrem às vezes, tanto em meninos quanto em meninas, mas isso não significa que a criança seja hermafrodita. A patologia é conhecida na prática médica mundial sob nomes diferentes: hiperplasia congênita (disfunção) do córtex adrenal ou síndrome de Aper-Gama, mas os especialistas nacionais preferem a definição mais tradicional de “síndrome adrenogenital em crianças”, embora não seja totalmente correta. O que é isso: uma piada triste da natureza, uma patologia genética comum ou um fenômeno médico raro que é quase impossível de encontrar na vida real? Vamos descobrir isso juntos.
O corpo humano é um sistema extremamente complexo, portanto a falha de um nó muitas vezes leva a sérios problemas em todo o “mecanismo”. Então, se por um motivo ou outro o funcionamento das glândulas supra-renais (mais precisamente, seu córtex) for perturbado, as consequências podem ser muito tristes, pois essas glândulas endócrinas são responsáveis pela produção de muitos hormônios que regulam o funcionamento de todos os órgãos. e sistemas do corpo. A síndrome adrenogenital é caracterizada por aumento da secreção de andrógenos e diminuição significativa dos níveis de aldosterona e cortisol. Em outras palavras, no corpo de um recém-nascido (isso se aplica igualmente a meninos e meninas) há muitos hormônios masculinos e hormônios femininos insignificantes.
Não é difícil adivinhar aonde isso pode levar. Sobre sintomas e manifestações clínicas falaremos um pouco mais tarde, mas de qualquer forma, seria um grande erro chamar um bebê assim de defeito. Ao contrário de outros adultos, cujo sexo é adivinhado com grande dificuldade, uma criança com doença congênita patologia genética definitivamente não é culpado de nada. Porém, a inércia do pensamento e o péssimo hábito de colocar etiquetas em tudo são uma força terrível, por isso os recém-nascidos do “sexo médio” (só o termo já vale a pena!) não terão o destino mais agradável. Mas com a abordagem correcta (e sobretudo isto diz respeito Equipe médica) tal cenário pode ser completamente evitado. O diagnóstico correto e oportuno, o tratamento adequado e uma abordagem individual (!) de cada paciente reduzirão significativamente a probabilidade de uma criança se tornar um pária.
Em trabalhos científicos conceituados, a síndrome adrenogenital (AGS) é convencionalmente dividida em três tipos: perdedora de sal, viril simples e neoclássica (pós-púbere). Eles diferem bastante, portanto outra má prática de cuidados de saúde domésticos - o diagnóstico apenas com base em diversas manifestações clínicas - é completamente inaceitável.
AGS é explicada apenas pela manifestação patologia hereditária, portanto é impossível “ficar doente” no sentido usual da palavra. Na maioria das vezes, ela se manifesta em recém-nascidos, mas às vezes os pacientes não percebem seu problema até os 30-35 anos de idade. EM o último caso Fatores que não são óbvios à primeira vista podem “desencadear” o mecanismo: aumento da radiação de fundo, tratamento com medicamentos potentes ou efeito colateral do uso de anticoncepcionais hormonais.
A síndrome adrenogenital é transmitida de forma autossômica recessiva, a partir da qual é possível prever a probabilidade de desenvolver patologia:
Alguns podem achá-los engraçados ou indecentes, mas, felizmente, quase não existem mais tais “únicos” agora. AGS não pode ser chamado de fatal patologia perigosa, mas alguns sintomas podem causar muitos momentos desagradáveis ou até mesmo levar a um colapso nervoso. Se a AGS for detectada em um recém-nascido, os pais terão tempo para ajudar a criança na adaptação social. Mas se um aluno for diagnosticado, a situação muitas vezes fica fora de controle, o que em alguns casos pode levar às consequências mais imprevisíveis. As próprias manifestações clínicas são as seguintes:
Além disso, os “meninos” são caracterizados por um pênis desproporcionalmente grande, e os pacientes com a forma não clássica de AGS muitas vezes se queixam de problemas de concepção e gravidez, razão pela qual praticamente não têm chance de se tornarem mães (na ausência de condições adequadas tratamento).
Como já dissemos, reconhecer o AGS é bastante difícil. A abordagem clássica ao diagnóstico, que envolve a recolha de uma história detalhada, a análise de doenças passadas e uma conversa detalhada com o paciente, revela-se insustentável. A razão para esta situação reside na idade da maioria dos pacientes (recém-nascidos, crianças em idade escolar e adolescentes). E dada a abordagem formal dos exames médicos escolares, não é surpreendente que muitas pessoas tomem conhecimento do seu diagnóstico quando adultos.
E se você “esquecer” por um tempo o exame médico inicial, eficaz e ao mesmo tempo métodos universais Restam apenas duas coisas para reconhecer o AGS:
Um exame aprofundado pode ser necessário para confirmar o diagnóstico. pesquisa genética, mas o custo desse procedimento não permite recomendá-lo como método diagnóstico universal.
AGS não é uma patologia fatal, portanto podemos falar sobre a probabilidade de desenvolvimento de alterações irreversíveis no organismo que podem levar a resultado fatal, felizmente, não é necessário. Mas se você perguntar se existem atualmente métodos eficazes para o tratamento da síndrome adrenogenital, a resposta será negativa. Pacientes com AGS são forçados a passar a vida inteira em terapia de reposição hormonal, o que lhes permite compensar a deficiência de hormônios glicocorticosteróides.
As perspectivas de longo prazo para tal tratamento não foram totalmente estudadas, mas há evidências que indicam uma alta probabilidade de desenvolver patologias cardiovasculares, doenças do trato gastrointestinal, bem como Neoplasias malignas. Mas, neste caso, o benefício do tratamento supera significativamente o risco potencial.
Atualmente, estão em andamento pesquisas para tratar a HAS por meio de transplante adrenal, mas ainda não saíram da fase experimental. Quando a tecnologia é desenvolvida, podemos assumir um custo extremamente elevado da operação e um risco considerável de efeitos colaterais.
Quanto métodos eficazes, que permitiriam “segurar” contra ASG não existem. Isso se explica pelo fato de sua ocorrência ser influenciada não por fatores de risco internos ou externos, mas sim por predisposição genética. Consequentemente, o único método de prevenção possível é o planejamento de uma futura gravidez com análise genética obrigatória de ambos os cônjuges. O custo de tal estudo é bastante elevado, por isso não pode ser recomendado a todos os casais sem levar em conta as capacidades financeiras.
A exposição à radiação ionizante e o envenenamento por substâncias tóxicas potentes também devem ser reconhecidos como fatores que podem desencadear a ativação de um gene defeituoso. Ao mesmo tempo, não há necessidade de falar em prevenção, mas durante o período de concepção e nascimento de um filho, é ainda melhor abster-se de visitar locais de testes nucleares e de caminhar por áreas com situação ambiental deprimente.
Geralmente tentamos abster-nos de qualquer conselho que implique a adesão cega a um ou outro dogma. E isso não é de forma alguma uma questão de atitude arrogante para com os leitores. Pelo contrário: não consideramos a nossa opinião a única correta, por isso preferimos dar-lhe a totalidade informação necessária, e não receitas generalizadas para todas as ocasiões. Mas a AGS é um caso especial.
Não vamos lembrar mais uma vez que uma criança que recebeu um diagnóstico “desagradável” não é um pária ou um urso de circo criado para divertir o público. E em qualquer caso, você o amará e o protegerá, embora não deva focar mais uma vez no gênero. Por outro lado, uma tutela superpróxima pode prestar-lhe um desserviço, e uma desculpa como “ele é incomum, pode estar ofendido” é apenas uma tentativa de protegê-lo do mundo exterior, fadado ao fracasso. Ajudar e apoiar é uma coisa, mas viver a vida para ele, transformar uma pessoa viva em manequim, é completamente diferente.

Icterícia fisiológica tratamento recém-nascido
A síndrome adrenogenital (SAG) é violação grave, ameaçando tanto a sua paz de espírito, saúde e vida, como a condição dos seus filhos. Afeta recém-nascidos, adolescentes e adultos de ambos os sexos. Por isso, é importante saber quais são os sintomas, formas, métodos de tratamento e prevenção desta doença. Neste artigo consideraremos todos esses aspectos e daremos recomendações sobre a prevenção do risco de AGS em bebês.
A síndrome adrenogenital (AGS) é um distúrbio grave que ameaça sua paz de espírito, saúde e vida
A síndrome adrenogenital é uma doença hereditária do córtex adrenal. Causa problemas cosméticos, físicos e psicológicos.
A patologia é de natureza congênita (hereditária) e é acompanhada por um distúrbio nos processos de síntese hormonal no córtex adrenal. Nesse caso, é produzida uma quantidade excessiva de andrógeno, o hormônio sexual masculino. Como resultado desse processo, observa-se a virilização (aparecimento ou exacerbação de traços masculinos em homens e mulheres).
A patologia é causada por mutações genéticas hereditárias que levam a um distúrbio do sistema enzimático adrenal. O que os pais devem esperar:
A patogênese (processos que ocorrem durante a doença) da síndrome adrenogenital consiste na produção excessiva do hormônio andrógeno devido à deficiência de uma determinada enzima. Ao mesmo tempo, a produção de outros hormônios (cortisol, que estimula a síntese protéica, e aldosterona, responsável pelo metabolismo dos minerais no organismo) é anormalmente reduzida. O grau da doença é determinado pela intensidade de secreção (produção) do excesso de substâncias.

Hormônio andrógeno
Existem várias formas de AGS congênita, com sintomas diferentes:
Às vezes, esse defeito hormonal pode não ser congênito, mas adquirido (forma pós-puberal). Nesse caso, ela se desenvolve em decorrência do aldosteroma, tumor que surge no córtex adrenal.

Forma hipertensiva de síndrome adrenogenital
Acima listamos os sintomas característicos de formas diferentes AGS hereditária. É preciso agilizar o que foi dito, e também acrescentar à lista de características indicadas.
Para tipo simples A forma viril da patologia é caracterizada por sintomas gerais:
Em crianças do sexo feminino, a síndrome adrenogenital apresenta as seguintes manifestações:

Sintomas da síndrome adrenogenital
Nos meninos, a síndrome adrenogenital é acompanhada pelos seguintes sintomas:
A forma pós-puberal ou não clássica (adquirida) de AGS é caracterizada pelos seguintes sintomas:
Para saber mais sobre os sinais mais comuns da AGS, você pode ver fotos que ilustram os sintomas da virilização.
AGS é diagnosticado examinando o paciente com vários médicos, incluindo:

Você precisa ser examinado por um endocrinologista
O médico faz a anamnese da doença e analisa as queixas do paciente. Ele examina um paciente em potencial para detectar anormalidades físicas primárias que indicam AGS.
É também fornecido um conjunto de estudos cujo objetivo é refutar doenças que apresentam sinais semelhantes aos da SAG. Esses estudos incluem:

Ultrassonografia pélvica
As características do combate a esse defeito são as seguintes:
O grau de cura depende se o diagnóstico for feito em tempo hábil. O diagnóstico precoce pode prevenir alterações genitais em meninas. Com a abordagem terapêutica correta das formas clássicas da doença na mulher, é possível garantir o funcionamento do parto e o curso normal do processo gravídico.
Como Medidas preventivas Os médicos aconselham recorrer a:
A síndrome adrenogenital é uma doença grave que ameaça ter consequências graves para mulheres e homens. Conhecer as características desta doença irá ajudá-lo a evitar as suas complicações: infertilidade, problemas estéticos excessivos e, em alguns casos, a morte.
Se você planeja ter filhos, deve monitorar cuidadosamente seus próprios indicadores de saúde. Você definitivamente deve consultar seu médico.
A síndrome adrenogenital (AGS) é uma fermentopatia hereditária com hiperplasia congênita do córtex adrenal. A patologia é baseada em uma interrupção geneticamente determinada do processo de esteroidogênese. A AGS é caracterizada por hipersecreção de andrógenos pelas glândulas supra-renais, supressão da produção de hormônios gonadotrópicos e glicocorticóides e comprometimento da foliculogênese.
EM medicamento oficial AGS é chamada de síndrome de Apère-Gamay. É caracterizada por desequilíbrio hormonal no corpo: níveis excessivos de andrógenos no sangue E quantidades insuficientes de cortisol e aldosterona. As consequências da doença são mais perigosas para os recém-nascidos. Seu corpo fica cheio de andrógenos e com baixo teor de estrogênio - hormônios sexuais masculinos e femininos.
Os primeiros sinais clínicos da doença aparecem nas crianças imediatamente após o nascimento. Em alguns, extremamente em casos raros, a AGS é detectada em indivíduos com idade entre 20 e 30 anos. A prevalência da síndrome varia significativamente entre os grupos étnicos: é mais elevada entre judeus, esquimós e caucasianos.
As glândulas supra-renais são glândulas endócrinas emparelhadas localizadas acima da parte superior dos rins humanos. Este órgão garante o funcionamento coordenado de todos os sistemas do corpo e regula o metabolismo. As glândulas supra-renais, juntamente com o sistema hipotálamo-hipófise, fornecem regulação hormonal de funções vitais. funções importantes corpo.
As glândulas supra-renais estão localizadas no retroperitônio e consistem em um córtex externo e uma medula interna. As células do córtex secretam glicocorticosteróides e hormônios sexuais. Os hormônios corticosteróides regulam o metabolismo e a energia, fornecem a defesa imunológica do corpo, tonificam parede vascular, ajuda a se adaptar ao estresse. A medula produz catecolaminas, substâncias biologicamente ativas.

O cortisol é um hormônio do grupo dos glicocorticosteróides secretado pela camada externa das glândulas supra-renais. O cortisol regula o metabolismo dos carboidratos e a pressão arterial, protege o corpo dos efeitos de situações estressantes, tem um leve efeito antiinflamatório e aumenta o nível de defesa imunológica.
A aldosterona é o principal mineralocorticóide produzido pelas células glandulares do córtex adrenal e regula o metabolismo do sal de água no corpo. Remove o excesso de água e sódio dos tecidos para o espaço intracelular, evitando a formação de edema. Ao atuar nas células renais, a aldosterona pode aumentar o volume sanguíneo circulante e aumentar a pressão arterial.
Existem 3 formas clínicas AGS, que se baseiam em vários graus de deficiência de 21-hidroxilase:

21-hidroxilase
AGS ocorre em indivíduos com deficiência congênita da enzima C21-hidroxilase. Para que sua quantidade no organismo seja mantida em um nível ideal, é necessário um gene completo, localizado nos autossomos do 6º cromossomo. Uma mutação deste gene leva ao desenvolvimento de uma patologia - um aumento no tamanho e uma deterioração no funcionamento do córtex adrenal.
A síndrome é herdada de forma autossômica recessiva - de ambos os pais ao mesmo tempo. No portador de um gene mutante, a síndrome não se manifesta clinicamente. A manifestação da doença só é possível na presença de genes defeituosos em ambos os autossomos do 6º par.
Padrões de transmissão hereditária da síndrome adrenogenital:

Em casos extremamente raros, a síndrome adrenogenital é herdada esporadicamente. O início repentino da patologia é devido a impacto negativo sobre o processo de formação de células germinativas femininas ou masculinas. Em casos extremamente raros, crianças doentes nascem de pais completamente saudáveis. A causa de tais anomalias pode ser neoplasias das glândulas supra-renais e processos hiperplásicos nas glândulas.
Ligações patogenéticas de AGS:
Fatores de risco que ativam o mecanismo da patologia:
As causas da AGS são exclusivamente de natureza hereditária, apesar da influência de fatores provocadores.
Principais sintomas da AGS:

A forma perdedora de sal é caracterizada por um curso grave e é rara. A doença se manifesta:
A forma perdedora de sal é caracterizada por hipercalemia, hiponatremia e hipocloremia.
A forma simples de AGS em meninos de 2 anos se manifesta:

A forma pós-puberal se manifesta em adolescentes:
Abortos, abortos espontâneos e gestações não desenvolvidas podem provocar o desenvolvimento desta forma de AGS.
Nas meninas, a forma viril clássica da AGS se manifesta pela estrutura intersexual da genitália externa: um clitóris grande e uma extensão da abertura uretral em sua cabeça. Os grandes lábios lembram o escroto, em axilas e os pelos pubianos começam a crescer cedo, músculos esqueléticos estão se desenvolvendo rapidamente. A AGS pronunciada nem sempre permite determinar o sexo do recém-nascido. As meninas doentes são muito parecidas com os meninos. As glândulas mamárias não crescem, a menstruação está ausente ou torna-se irregular.

Crianças com AGS são monitoradas por endocrinologistas pediátricos. Usando técnicas terapêuticas modernas, os especialistas fornecem medicamentos e cirurgia síndrome, que permite que o corpo da criança se desenvolva corretamente no futuro.
AGS não é fatal doença perigosa, embora alguns de seus sintomas deprimam psicologicamente os pacientes, o que muitas vezes termina em depressão ou colapso nervoso. A detecção oportuna de patologia em recém-nascidos permite que crianças doentes se adaptem à sociedade ao longo do tempo. Quando a doença é detectada em crianças em idade escolar, a situação muitas vezes fica fora de controle.
O diagnóstico da SAG é baseado em dados anamnésicos e fenotípicos, bem como nos resultados pesquisa hormonal. Durante um exame geral, são avaliados a figura do paciente, a altura, a condição dos órgãos genitais e o grau de crescimento do cabelo.

Diagnóstico laboratorial:
Diagnóstico instrumental:
A triagem neonatal é realizada no 4º dia após o nascimento da criança. Uma gota de sangue é retirada do calcanhar do recém-nascido e aplicada em uma tira-teste. As demais táticas de manejo de uma criança doente dependem dos resultados obtidos.
AGS requer tratamento hormonal vitalício. Para as mulheres adultas, a terapia de reposição é necessária para a feminização, para os homens é realizada para eliminar a esterilidade e para as crianças para superar as dificuldades psicológicas associadas ao desenvolvimento precoce das características sexuais secundárias.
A terapia medicamentosa para a doença consiste no uso dos seguintes medicamentos hormonais: 
As crises de insuficiência adrenal podem ser evitadas aumentando a dose de corticosteróides em 3-5 vezes. O tratamento é considerado eficaz se o ciclo menstrual da mulher tiver normalizado, a ovulação tiver ocorrido e a gravidez tiver ocorrido.
O tratamento cirúrgico da AHS é realizado em meninas de 4 a 6 anos. Consiste na correção da genitália externa – cirurgia plástica vaginal, clitoridectomia. A psicoterapia é indicada para aqueles pacientes que não conseguem se adaptar de forma independente à sociedade e não se percebem como uma pessoa plena.
Se houver histórico familiar de hiperplasia adrenal, todos os casais precisam consultar um geneticista. Diagnóstico pré-natal consiste no monitoramento dinâmico de uma gestante de risco por 2 a 3 meses.
A prevenção de AGS inclui:
O diagnóstico oportuno e a terapia de reposição de alta qualidade tornam o prognóstico da doença relativamente favorável. O tratamento hormonal precoce estimula desenvolvimento adequadoórgãos genitais e permite preservar função reprodutiva em mulheres e homens.
Se o hiperandrogenismo persistir ou não for corrigível com corticosteróides, os pacientes permanecerão com baixa estatura e apresentarão características defeitos cosméticos. Isso perturba a adaptação psicossocial e pode levar a um colapso nervoso. O tratamento adequado permite que mulheres com formas clássicas de AGS engravidem, tenham e dêem à luz uma criança saudável.
Doença hereditária das glândulas supra-renais, na qual a esteroidogênese é prejudicada devido à falha funcional das enzimas. Manifesta-se por virilização dos órgãos genitais, físico masculino, subdesenvolvimento mamário, hirsutismo, acne, amenorreia ou oligomenorreia, infertilidade. Durante o diagnóstico, são determinados os níveis de 17-hidroxiprogesterona, 17-cetosteroides, androstenediona, ACTH e é realizada uma ultrassonografia dos ovários. Os pacientes recebem terapia de reposição hormonal com glicocorticóides e mineralocorticóides, estrogênios em combinação com andrógenos ou progestágenos de nova geração. Se necessário, é realizada cirurgia plástica genital.

A síndrome adrenogenital, ou disfunção congênita (hiperplasia) do córtex adrenal, é a doença hereditária mais comum. A prevalência da patologia difere entre representantes de diferentes nacionalidades. Opções clássicas AGS em caucasianos ocorre com uma frequência de 1:14.000 crianças, enquanto nos esquimós do Alasca esse número é de 1:282. A incidência da doença é significativamente maior entre os judeus. Assim, a forma não clássica do distúrbio adrenogenital é detectada em 19% dos indivíduos Nacionalidade judaica Grupos Ashkenazi. A patologia é transmitida de forma autossômica recessiva. A probabilidade de ter um filho com essa síndrome quando ambos os pais são portadores do gene patológico chega a 25%, no casamento entre portador e paciente - 75%. Se um dos pais tiver DNA completo, as manifestações clínicas da síndrome não se desenvolvem nas crianças. Se o pai e a mãe tiverem SDA, a criança também ficará doente.
Nas formas pré-natais da doença (viril simples e perdedora de sal), o principal sintoma clínico é a virilização visível dos órgãos genitais. As meninas recém-nascidas apresentam sinais de pseudo-hermafroditismo feminino. O clitóris é grande ou em forma de pênis, o vestíbulo da vagina é aprofundado, o seio urogenital é formado, os grandes e pequenos lábios são aumentados e o períneo é alto. Os órgãos genitais internos são desenvolvidos normalmente. Em meninos, o pênis está aumentado e o escroto hiperpigmentado. Além disso, no distúrbio adrenogenital perdedor de sal, os sintomas de insuficiência adrenal são expressos em distúrbios somáticos graves, muitas vezes incompatíveis com a vida (diarréia, vômito, convulsões, desidratação, etc.), que aparecem a partir de 2 a 3 semanas de idade.
Nas meninas com AGS viril simples, à medida que envelhecem, os sinais de virilização se intensificam e um físico displásico é formado. Devido à aceleração dos processos de ossificação, os pacientes apresentam baixa estatura, ombros largos, pelve estreita e membros curtos. Ossos tubulares enorme. Puberdade começa cedo (antes dos 7 anos) e prossegue com o desenvolvimento das características sexuais masculinas secundárias. Há aumento do clitóris, diminuição do timbre da voz, aumento da força muscular e formação de uma forma masculina típica da cartilagem cricóide da glândula tireóide. Os seios não crescem, a menarca está ausente.
Menos específicos são os sintomas clínicos das formas não clássicas da síndrome virilizante que ocorrem durante a puberdade e após o estresse (aborto espontâneo no início da gravidez, aborto medicamentoso, cirurgia, etc.). Normalmente, os pacientes lembram que, mesmo na idade escolar, começaram a apresentar ligeiro crescimento de pelos nas axilas e na região pubiana. Posteriormente, surgiram sinais de hirsutismo com o crescimento de fios de cabelo acima do lábio superior, ao longo da linha branca do abdômen, no esterno e na região areolopapilar. Mulheres com AGS queixam-se de acne persistente, porosidade e aumento da oleosidade da pele.
A menarca chega tarde - por volta dos 15-16 anos. O ciclo menstrual é instável, os intervalos entre as menstruações chegam a 35-45 dias ou mais. A secreção sanguinolenta durante a menstruação é escassa. As glândulas mamárias são pequenas. O clitóris está ligeiramente aumentado. Essas meninas e mulheres podem ser altas, ter pélvis estreita e ombros largos. De acordo com observações de especialistas da área de obstetrícia e ginecologia, quanto mais tarde se desenvolvem os distúrbios adrenogenitais, menos perceptíveis são os sinais externos característicos dos homens e mais frequentemente o principal sintoma se torna uma violação do ciclo menstrual. Com defeitos genéticos mais raros, os pacientes podem queixar-se de aumento da pressão arterial ou, inversamente, hipotensão com baixo desempenho e dores de cabeça frequentes, hiperpigmentação da pele com sintomas mínimos de virilização.
A principal complicação da síndrome adrenogenital, para a qual os pacientes recorrem ao obstetra-ginecologista, é a infertilidade persistente. Quanto mais cedo a doença se manifestar, menor será a probabilidade de engravidar. Com deficiência enzimática significativa e manifestações clínicas de síndrome virilizante simples, a gravidez não ocorre. Pacientes grávidas com formas puberais e pós-puberais da doença apresentam abortos espontâneos nos estágios iniciais. Durante o parto, é possível a insuficiência ístmico-cervical funcional. Essas mulheres são mais propensas à ocorrência de distúrbios psicoemocionais - tendência à depressão, comportamento suicida e manifestações de agressão.
Fazendo um diagnóstico para tipos pré-natais de AGS com mudanças característicasórgãos genitais não é difícil e é realizado imediatamente após o parto. Em casos duvidosos, o cariótipo é utilizado para confirmar o cariótipo feminino (46XX). A busca diagnóstica torna-se mais importante nos casos de início clínico tardio ou curso latente com mínima manifestações externas virilização. EM situações semelhantes Para identificar a síndrome adrenogenital, são utilizados os seguintes métodos laboratoriais e instrumentais:
A variante perdedora de sal da AGS também é caracterizada por um aumento da concentração de renina no plasma sanguíneo. O diagnóstico diferencial dos distúrbios adrenogenitais que surgem durante a puberdade e idade fértil é realizado com síndrome dos ovários policísticos, androblastomas ovarianos, androsteromas adrenais, síndrome viril de origem hipotalâmica e hirsutismo constitucional. Em casos difíceis, endocrinologistas, urologistas e geneticistas estão envolvidos no diagnóstico.
A principal forma de corrigir a disfunção adrenal viril é a reposição terapia hormonal, reabastecendo a deficiência de glicocorticóides. Se uma mulher com AGS latente não tiver planos reprodutivos, manifestações cutâneas o hiperandrogenismo é insignificante e a menstruação é rítmica, não são utilizados hormônios. Em outros casos, a escolha do regime de tratamento depende da forma da patologia endócrina, dos principais sintomas e do grau de sua gravidade. Muitas vezes, a prescrição de glicocorticóides é complementada com outros métodos médicos e cirúrgicos, selecionados de acordo com o objetivo terapêutico específico:
Certas dificuldades no manejo da paciente surgem nos casos em que a doença não é diagnosticada em hospital obstétrico e uma menina com virilização grave dos órgãos genitais é registrada e criada como menino. Na decisão de restaurar a identidade de gênero feminina, a cirurgia plástica cirúrgica e a terapia hormonal são complementadas com apoio psicoterapêutico. A decisão de preservar o sexo masculino civil e remover o útero e anexos é tomada em casos excepcionais por insistência dos pacientes, mas esta abordagem é considerada errônea.
O prognóstico para detecção oportuna da síndrome adrenogenital e terapia adequadamente selecionada é favorável. Mesmo em pacientes com virilização significativa dos órgãos genitais após cirurgia plástica, normal vida sexual e parto natural. A terapia de reposição hormonal para qualquer forma de AGS promove rápida feminização - desenvolvimento glândulas mamárias, aparecimento da menstruação, normalização do ciclo ovariano, restauração da função geradora. A prevenção da doença é feita na fase de planejamento da gravidez.
Caso tenham sido observados casos dessa patologia na família, está indicada a consulta com um geneticista. A realização de um teste de ACTH para ambos os cônjuges permite diagnosticar portadores heterozigotos ou formas latentes de distúrbio adrenogenital. Durante a gravidez, a síndrome pode ser detectada pelos resultados de uma análise genética das células das vilosidades coriônicas ou do conteúdo do líquido amniótico obtido por amniocentese. A triagem neonatal, realizada no 5º dia após o nascimento, tem como objetivo identificar concentrações aumentadas de 17-hidroprogesterona para rápida seleção de táticas terapêuticas.

O sentido da vida está relacionado com a questão “Para que viver”, e não com a questão de como manter a vida. A atitude de uma pessoa é...

Um cogumelo é um organismo vivo que forma um reino separado com o mesmo nome. Por muito tempo foram classificados como parte do reino vegetal. Mas em...

Para os amantes da caça “tranquila”, a temporada dos cogumelos começa no início do verão e vai até o final do outono. E raramente o fazem...

Aleshnikova, V.I. Uso de consultores profissionais. - M.: Infra-M, 1999. - 240 p. 2. Beich, E....

Suco de laranja. O significado simbólico do suco de laranja nos livros de sonhos é prazer e tentação. Muitas vezes nós...

Na maioria das vezes superamos todos os tipos de dificuldades encontradas ao longo do caminho da vida. Claro, para isso fazemos...

Este ano o seu patrono Netuno estará na sua constelação e isso é um bom sinal, pois você...

1993 quem? 1993 é o ano de qual animal? — De acordo com o horóscopo chinês, 1873, 1933, 1993 pertenciam aos anos do Negro...

A dualidade onda-partícula da luz significa que a luz tem simultaneamente as propriedades de continuidade...

O papel da biologia é enorme em nosso mundo. Embora não seja uma das disciplinas prioritárias, a maioria dos escolares e...
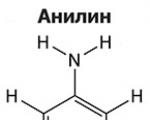
As aminas são derivados orgânicos da amônia contendo um grupo amino NH 2 e um radical orgânico. Em geral...
Como responder às perguntas da Parte BA segunda parte do trabalho de estudos sociais consiste em 7 tarefas com uma resposta curta....

O formulário TORG-15 é elaborado no caso de durante o transporte, movimentação entre e dentro do armazém, quando...

Os nutricionistas dizem que para ter uma boa saúde e um corpo esguio é preciso incluir lanches na sua...

Um cogumelo é um organismo vivo que forma um reino separado com o mesmo nome. Durante muito tempo foram classificados como um reino...

Para os amantes da caça “tranquila”, a temporada dos cogumelos começa no início do verão e vai até o final do outono. E raramente o fazem...