Що можна і не можна на Різдвяний піст?
У 2018 році Різдвяний пост розпочнеться 28 листопада. У цей період православні віруючі готуються до зустрічі Різдва.
Дистопія - це опущення мигдаликів мозочка у великий потиличний отвір і спинномозковий канал. Також цю аномалію називають мальформацією Кіарі І ступеня.
Зазвичай даний діагнозне викликає будь-яких значних порушень та занепокоєння пацієнта. Найчастіше проявляється у віці 30 – 40 років. Під час досліджень є випадковою «знахідкою».
Найчастіше – це вроджена патологія, що проявляється невідповідністю розмірів головного мозку та потиличного отвору Дистопія мозочка може бути і вторинною – при частих та травматичних люмбальних пункціях.

Симптом дистопії рідко проявляється клінічно. Однак можлива поява і неврологічних ознак- Болі в шиї при напруженні, кашлі. Вона носить «стріляючий» і нападоподібний характер. Виникають головні болі, запаморочення. Чим більше явище дистопії (варіюється від кількох міліметрів до кількох сантиметрів), тим більше виражаються розлади. При сильному опущенні може виникнути сирингомієлія (розширення каналу спинного мозкута утворення порожнин навколо нього).
При негативних неврологічних симптомах лікування зазвичай не потрібне. Пацієнтів з мінімальною чи відсутньою симптоматикою все одно залишають під наглядом лікаря-невролога для моніторингу розвитку захворювання.
Якщо неврологічна симптоматика все ж таки є, але в незначних проявах – можливе консервативне лікування. Купірування болів судинними препаратамита нестероїдними протизапальними ліками, міорелаксантами. Також необхідні рекомендації щодо способу життя і режиму.

Хірургічне втручання – єдине ефективний виглядлікування для хворих із широкими розладами.
Хірургічне лікування передбачає розширення черепної ямкита пластики твердої мозкової оболонки.
Основною методикою для діагностики є магнітно-резонансна томографія. Комп'ютерна томографія та рентгенівське дослідження не дадуть чіткої картини захворювання.
Якщо у вас при МРТ виявили дистопію мигдаликів мозочка, не варто переживати з цього приводу. Найчастіше аномалія відбувається безсимптомно і вимагає лікування.
Аномалія Арнольда-Кіарі I типу (Q07.0) - це вроджена аномаліярозвитку головного мозку, що характеризується усуненням утворень задньої черепної ямки в область хребетного каналу нижче рівня великого потиличного отвору.
Поширеність: загалом 3,3-8,2 на 100 тисяч жителів. Дебют клінічних симптомів спостерігається у дорослому або підлітковому віці. У 50-75% випадків відзначається поєднання із сирингоміелією, у 10% - з гідроцефалією.
Іноді дана патологіяне має клінічних симптомів, стає рентгенологічною знахідкою.
Основні симптоми аномалії Арнольда-Кіарі І типу такі:
При об'єктивному огляді пацієнта можна виявити загальномозковий синдром, платибазію (50%), ністагм, диплопію, дисфагію, дизартрію, гіпестезю в ділянці шиї, рук, спастичні парези в руках, пожвавлення глибоких рефлексів, атаксію.
Диференціальний діагноз:
Лікування – симптоматичне. Призначається лише після підтвердження діагнозу лікарем-фахівцем. Наростання клінічної симптоматики- Показання для субокципітальної декомпресії, шунтуючих операцій.
Є протипоказання. Необхідна консультація спеціаліста.





(мальформація Арнольда-Кіарі) - захворювання, при якому структури головного мозку, розташовані в задній черепній ямці, опущені в каутальному напрямку і виходять через великий потиличний отвір. Залежно від типу аномалія Кіарі може виявлятися головним болем у потилиці, болем у шийному відділі, запамороченням, ністагмом, непритомністю, дизартрією, мозочковою атаксією, парезом гортані, зниженням слуху та вушним шумом, порушенням зору, дисфагією, дихальними апное, стридором, розладами чутливості, гіпотрофією м'язів та тетрапарезом. Аномалія Кіарі діагностується шляхом проведення МРТ головного мозку, шийного та грудного відділівхребта. Аномалія Кіарі, що супроводжується стійким больовим синдромом або неврологічним дефіцитом, підлягає хірургічному лікуванню (декомпресія задньої черепної ямки або операції, що шунтують).

В області з'єднання черепа з хребетним стовпомзнаходиться великий потиличний отвір, на рівні якого стовбур головного мозку переходить у спинний мозок. Вище цього отвору локалізується задня черепна ямка. У ній розташований міст, довгастий мозок і мозок. Аномалія Кіарі пов'язана з виходом частини анатомічних структур задньої черепної ямки у просвіт великого потиличного отвору. При цьому відбувається здавлення структур, що знаходяться в цій галузі, довгастого і спинного мозку, а також порушення відтоку цереброспінальної рідини з головного мозку, що призводить до гідроцефалії. Разом з платибазією, асиміляцією атланту та ін. аномалія Кіарі відноситься до вроджених вад розвитку краніовертебрального переходу.
Аномалія Кіарі зустрічається за різними даними у 3-8 осіб на 100 тисяч населення. Залежно від типу аномалія Кіарі може діагностуватись у перші дні після народження дитини або стати несподіваною знахідкою у дорослого пацієнта. У 80% випадків аномалія Кіарі поєднується із сирингомієлією.
Досі аномалія Кіарі залишається захворюванням, про етіологію якого в неврології немає єдиної думки. Ряд авторів вважає, що аномалія Кіарі пов'язана зі зменшеним розміром задньої черепної ямки, що призводить до того, що зі зростанням розташованих у ній структур вони починають виходити через потиличний отвір. Інші дослідники припускають, що аномалія Кіарі розвивається в результаті збільшених розмірів головного мозку, який при цьому ніби виштовхує вміст задньої черепної ямки через потиличний отвір.
Спровокувати перехід незначно вираженої аномалії у виражену клінічну формуможе гідроцефалія, при якій за рахунок збільшення шлуночків збільшується загальний обсяг мозку. Оскільки аномалія Кіарі поряд з дисплазією кісткових структур краніовертебрального переходу супроводжується недорозвиненням зв'язкового апарату цієї області, будь-яка черепно-мозкова травма може призводити до погіршення вклинення мигдаликів мозочка в потиличний отвір з маніфестацією. клінічної картинизахворювання.
Аномалія Кіарі поділяється на 4 типи:
Характеризується опущенням мигдаликів мозочка нижче великого потиличного отвору. Зазвичай вона проявляється у підлітків чи дорослому віці. Найчастіше супроводжується гідроміелією - скупченням цереброспінальної рідини у центральному каналі спинного мозку.
Виявляється у перші дні після народження. Крім мигдаликів мозочка при цій патології через великий потиличний отвір виходять також черв'як мозочка, довгастий мозок і IV шлуночок. Аномалія Кіарі II типу набагато частіше поєднується з гідроміелією, ніж перший тип, і в переважній більшості випадків пов'язана з мієломенінгоцеле - уродженою спинномозковою грижею.
Відрізняється тим, що мозок, що опустилися через великий потиличний отвір і довгастий мозок, розташовуються в менінгоцелі шийно- потиличної області.
Полягає в гіпоплазії (недорозвитку) мозочка і не супроводжується його зміщенням у каутальному напрямку. Деякі автори відносять цю аномалію до синдрому Денді-Уокера, при якому гіпоплазія мозочка поєднується з наявністю вроджених кіст задньої черепної ямки та гідроцефалією.
Аномалія Кіарі II та Кіарі III часто спостерігається у комбінації з іншими дисплазіями нервової системи: гетеротопією кори головного мозку, полімікрогірією, аномаліями мозолистого тіла, кістами отвору Можанді, перегином сильвієвого водопроводу, гіпоплазією підкіркових структур, шатра та серпа мозочка.
Найчастіше в клінічній практицізустрічається аномалія Кіарі І типу. Вона проявляється лікворногіпертензійним, церебеллобульбарним та сирингомієлічним синдромами, а також ураженням черепно-мозкових нервів. Зазвичай аномалія Кіарі I маніфестує в період статевого дозрівання або вже у дорослому віці.
Для лікворногіпертензійного синдрому, яким супроводжується аномалія Кіарі I, характерний головний біль у потилиці та шийній ділянці, що посилюється під час чхання, кашлю, напруження або напруження м'язів шиї. Може спостерігатися блювання, яка залежить від прийому їжі та її характеру. Під час огляду пацієнтів з аномалією Кіарі виявляється підвищений тонусм'язів шиї. Серед мозочкових порушеньспостерігаються порушення мови (дизартрія), ністагм, мозочкова атаксія.
Ураження стовбура мозку, розташованих у ньому ядер черепно-мозкових нервів та їх корінців проявляються зниженням гостроти зору, диплопією, розладом ковтання, зниженням слуху за типом кохлеарного невриту, системним запамороченням з ілюзією обертання навколишніх предметів, вушним шумом, синдромом сонних апное свідомості, ортостатичним колапсом. Пацієнти, які мають аномалію Кіарі, відзначають посилення запаморочення і вушного шуму при поворотах головою. Поворот голови у таких хворих може спровокувати непритомність. Може відзначатися атрофічні зміни половини мови та парез гортані, що супроводжується осиплістю голосу та утрудненням дихання. Можливий тетрапарез з великим зниженням м'язової сили верхніх кінцівкахніж у нижніх.
У випадках, коли аномалія Кіарі I поєднується із сирингомієлією, спостерігається сирингомієлічний синдром: порушення чутливості за дисоційованим типом, оніміння, м'язові гіпотрофії, тазові порушення, нейроартропатії, зникнення черевних рефлексів. При цьому деякі автори вказують на невідповідність розміру та місцезнаходження сирингомієлічної кісти поширеності розладів чутливості, ступеня виразності парезів та м'язової гіпотрофії.
Аномалія Кіарі II та Кіарі III мають подібні клінічні прояви, які стають помітними з перших хвилин життя дитини. Аномалія Кіарі II супроводжується шумним диханням (вроджений стридор), періодами короткочасної зупинки дихання, двостороннім нейропатичним парезом гортані, порушенням ковтання із закиданням рідкої їжі в ніс. У новонароджених аномалія Кіарі II проявляється також ністагмом, підвищенням м'язового тонусуу верхніх кінцівках, ціанозом шкірних покривів, що виникають під час годування. Двигуниможуть бути виражені в різного ступеняі прогресувати до тетраплегії. Аномалія Кіарі III має важчий перебіг і найчастіше є несумісним із життям порушенням розвитку плода.
Неврологічний огляд та стандартний перелік неврологічних обстежень(ЕЕГ, Ехо-ЕГ, РЕГ) не дають специфічних даних, що дозволяють встановити діагноз «аномалія Кіарі». Як правило, вони виявляють лише ознаки значного підвищення внутрішньочерепного тиску, Т. е. гідроцефалію. Рентгенографія черепа виявляє лише кісткові аномалії, якими може супроводжуватися аномалія Кіарі. Тому до впровадження в неврологічну практику томографічних методів дослідження діагностика цього захворювання становила невролога великі труднощі. Наразі лікарі мають можливість поставити таким пацієнтам точний діагноз.
Слід зазначити, що МСКТ і КТ головного мозку при хорошій візуалізації кісткових структур краніовертебрального переходу не дозволяють досить точно судити про м'якоткані утворення задньої черепної ямки. Тому єдиним достовірним методом діагностики аномалії Кіарі на сьогоднішній день є магнітно-резонансна томографія. Її проведення вимагає знерухомленості пацієнта, тому у маленьких дітей вона проводиться у стані медикаментозного сну. Крім МРТ головного мозку для виявлення менінгоцеле та сирингомієлічних кіст необхідно також проведення МРТ хребта, особливо його шийного та грудного відділів. При цьому проведення МРТ досліджень має бути спрямоване не лише на діагностику аномалії Кіарі, а й на пошук інших аномалій розвитку нервової системи, які часто з нею поєднуються.
Безсимптомно протікає аномалія Кіарі не потребує лікування. У випадках, коли аномалія Кіарі проявляється лише наявністю болів у шиї та потиличній ділянці, проводять консервативну терапію, що включає аналгетичні, протизапальні та міорелаксуючі препарати. Якщо аномалія Кіарі супроводжується неврологічними порушеннями (парези, розлади чутливості і м'язового тонусу, порушення з боку черепно-мозкових нервів тощо) або больовим синдромом, що не піддається консервативної терапії, то показано її хірургічне лікування.
Найчастіше в лікуванні аномалії Кіарі застосовується краніовертебральна декомпресія. Операція включає розширення потиличного отвору за рахунок видалення частини потиличної кістки; ліквідацію здавлення стовбура та спинного мозку за рахунок резекції мигдаликів мозочка та задніх половин двох перших шийних хребців; нормалізацію циркуляції цереброспінальної рідини шляхом підшивання у тверду мозкову оболонкулатки зі штучних матеріалів або алотрансплантату. У деяких випадках аномалія Кіарі лікується за допомогою шунтуючих операцій, спрямованих на дренування цереброспінальної рідини із розширеного центрального каналу спинного мозку. Цереброспінальна рідина може відводитися в грудну або черевну порожнину(люмбоперитонеальне дренування).
Важливе прогностичне значення має тип, до якого належить аномалія Кіарі. У деяких випадках аномалія Кіарі I може протягом усього життя пацієнта зберігати безсимптомний перебіг. Аномалія Кіарі III у більшості випадків призводить до летального результату. З появою неврологічних симптоміваномалії Кіарі I, а також при аномалії Кіарі II велике значеннямає своєчасне проведення хірургічного лікування, оскільки неврологічний дефіцит погано відновлюється навіть після успішно проведеної операції. За різними даними, ефективність хірургічної краніовертебральної декомпресії становить 50-85%.
Як відомо, в організмі людини знаходиться 6 мигдалин. Кожна з них виконує важливу функцію. Вище великого потиличного отвору знаходяться мигдалики мозочка, тому вони розташовуються зазвичай. Ектопія мигдаликів - це найпоширеніша аномалія мозочка, а саме зміщення або розташування мигдаликів не на місці. Науково таку аномалію прийнято називати аномалією Арнольда-Кіарі.
Захворювання характеризується опущенням (одне або двостороннє) мигдаликів мозочка в хребетний канал через великий потиличний отвір. Важливим моментому такому разі є те, що якісь симптоми з'являються лише до тридцяти, а іноді й до сорока років. Якщо ектопія мигдаликів не супроводжується симптомами і виявляється випадково в результаті інших досліджень, то немає жодної необхідності її лікувати.
Питання про медикаментозне лікування або оперативне втручання виникає лише в тому випадку, якщо ектопія виявляє симптоми.
Причини цього захворювання поки що медициною не виявлено. Відомо лише, що має вплив генетичний спадок. При ектопії мигдаликів можуть спостерігатися наступні неврологічні синдроми. Хворий страждає
При ектопії мигдаликів є цілий ряд можливих порушень, Залежно від стадії її розвитку.
Раніше медицині потрібно більше часу і зусиль для діагностики ектопії мигдаликів, т. до. Комп'ютерна томографіядають не зовсім чітку картину. Тепер за допомогою МРТ можна отримати чітку картинку задньої черепної ямки, спинного мозку і т. д., на якій і буде видно порушення розташування мигдаликів мозочка.
Людям, яким було поставлено діагноз у результаті «випадкового» виявлення, не варто турбуватися. Хвороба ніяк не позначиться на вашому самопочутті чи тривалості життя. Якщо ж ви відчуваєте деякі симптоми ектопії мигдаликів, то варто все ж таки звернутися до лікаря, інакше можуть бути наслідки, наприклад, аномалія хребта.
Лікування хвороби в такому випадку може бути хірургічним та медикаментозним. Під час операції збільшують обсяг черепної ямки. Якщо з симптомів ектопії мигдаликів у вас присутній лише больовий синдром, можна обмежитися нестероїдними протизапальними препаратами, і операція у разі не потрібна.
Проте можливі такі її прояви:
Причин розвитку цієї аномалії достовірно невідомі.
Можливо, що ризик народження дитини з аномалією Арнольда-Кіарі збільшують такі фактори, що впливають на організм вагітної жінки:
До вродженим причинможе бути віднесено неправильний розвиток кісток черепа:
До набутих причин можуть бути віднесені:
LookMedBook нагадує: чим раніше Ви звернетеся за допомогою до фахівця, тим більше шансів зберегти здоров'я та знизити ризик розвитку ускладнень:
Діагноз ставиться виходячи з неврологічного огляду та результатів інструментального обстеження.
Обсяг та вид лікування залежать від ступеня тяжкості захворювання.
Через недостатність даних про причини захворювання заходи профілактики зводяться до правил здорового образужиття матері під час вагітності:
Передумовами до опущення мигдаликів є:
Опущення мигдаликів мозочка у великий потиличний отвір або кагал спинного мозку називають дистопією. А іноді ця патологія називається аномалією Кіарі. Як правило, таке захворювання не спричиняє значних розладів або явних симптомів і не має причин для занепокоєння хворого. Часто ця патологія проявляється після досягнення 30-40-річного віку. Виявляється зазвичай під час обстежень з інших причин. Тому про це захворювання слід знати, щоб воно не виявилося несподіванкою для хворого.
Для того, щоб зрозуміти, що таке низьке розташування мигдаликів мозочка, необхідно чітко знати клінічні симптомита як виявляється патологія. А оскільки цей стан супроводжується частими головними болями, потрібно спочатку встановити їхню причину, а потім уже займатися лікуванням. Таке захворювання виявляється при МРТ.
Дистопія мозочка, як правило, є вродженою патологією. Вона виникає при усуненні будь-якого органу в ембріональний період. Вторинне вона буває лише при проведенні частих пункцій або при люмбальних травмах. Інших причин появи даного захворюванняне виявлено.
Мигдалики мозочка дуже подібні до тих, які знаходяться в гортані. У нормальному становищівони розташовуються вище за БЗО черепа. А відхилення в їх розвитку та становищі можуть спричинити не лише дистопію. Найчастіше зустрічається опущення мигдаликів мозочка нижче рівня черепа.
До цього часу захворювання Кіарі є патологією, про причини виникнення якого неврологи не дійшли однієї думки. Деякі вважають, що ця аномалія виникає при зменшенні габаритів ямки позаду черепного вихідного отвору до спинного мозкового каналу. Це нерідко призводить до таких наслідків у процесі зростання тканин, які розташовані у коробці. Вони виходять у потиличний вихідний канал. Інші фахівці вважають, що захворювання починає розвиватися через збільшення обсягів мозкових тканин голови. У цьому випадку мозок починає виштовхувати через задню черепну ямку в потиличний черепний отвір мозочка і його мигдалики.
Викликає таке прогресування вираженої аномалії та її перехід у «клініку», як гідроцефалія. При цьому збільшуються загальні розміри мозку, особливо мозочкові тканини. Патологію Кіарі разом із недорозвиненим зв'язковим апаратом мозку супроводжує дисплазія тканин кістки. Тому кожна черепно-мозкова травма часто призводить до посилення зниження рівня знаходження мигдаликів і мозочка.
Є такі види аномальних відхилень – дистопія та аномалія Кіарі.
У свою чергу, захворювання Кіарі поділяють на чотири різні типи:
II та III типи часто проявляються у поєднанні з явищами дисплазії нервової системи, наприклад, з гетеротопією мозкових тканин кори, кістами отвору та ін.
Найпоширенішою серед аномалій є патологія першого виду. При ній нерідко можливий прояв лікворно-гіпертензійного синдрому, а також церебело-бульбарне та сирингомієлічне явища, порушення роботи нервових закінченьвсередині черепа.
Лікворно-гіпертензійний синдром – це біль у потилиці та шийних м'язах, які посилюються під час чхання, кашлю або напруги шийних м'язових тканин. Часто болі супроводжуються блюванням, не пов'язаним із прийомами їжі. Багато симптомів патології проявляються в залежності від того, в якому положенні розташовуються мигдалики мозочка щодо отвору, що знаходиться в потиличній ямці черепа. Спостерігається також:
Аномалія II та III типів має схожі симптоми, помітні вже з перших миттєвостей після народження малюка. Другий тип супроводжує гамірне дихання, а також несподівані напади зупинки дихання, нейропарез гортанних тканин. Спостерігаються відхилення процесу ковтання.
Ознаки дистопії рідко бувають очевидними. Але все ж таки можливі неврологічні прояви:
Якщо опущення мигдаликів сильне, іноді спостерігається розширення каналу, що зв'язує головний та спинний мозок, а також утворюються порожнини навколо каналу.
Основним сучасним методомдіагностування дистопії є МРТ. У цьому випадку ні КТ, ні рентгенівські дослідження не дають повної картинипатології.
Для діагностування синдрому Кіарі не підійдуть жодні стандартні методитипу ЕЕГ, ЕхоЕГ або РЕГ, тому що вони не дозволяють точно поставити діагноз. Огляд невролога також не визначить аномалію. Всі ці методи можуть показати підозру лише на підвищений тиск усередині черепної коробки. Рентгенографію черепа також варто робити, оскільки вона показує лише аномалії кісткових тканин, які можуть супроводжувати патологію. Тому до введення в діагностичну практику томографії діагностувати хворобу було проблематично. Сучасні способи діагностування дозволяють точно визначити патологію.
У разі якісної візуалізації кісткових тканин вертебрального переходу такі методи як МСКТ або КТ не дають достатньо точної картини. Єдиним достовірним способомдіагностування аномалії Кіарі на сьогодні є лише МРТ.
Оскільки проведення дослідження цим методом вимагає нерухомості хворого, маленьких дітей занурюють у штучний сон за допомогою ліків. Проводиться також МРТ спинного мозку. Вона спрямована на діагностику будь-яких аномальних відхилень у роботі нервової системи.
Консервативні методи лікування можливі лише за дуже незначних відхилень. Все залежить від того, який стан хворого на момент звернення до лікаря. У цьому випадку лікування спрямоване на зняття хворобливих симптомів нестероїдними лікамичи міорелаксантами. Необхідна також корекція режиму.
Єдиним ефективним методомлікування при широких відхиленнях є хірургічне втручання, яке полягає в розширенні ямки черепа та пластики твердих мозкових тканин оболонки.
Показаннями для оперативного лікування є:
Якщо аномальне відхилення протікає без відчутних ознак, лікування не потрібно. У випадках виникнення хворобливих відчуттівв районі шиї та потилиці проводиться консервативна терапія, при якій використовують аналгетики та асептичні лікарські речовини, а також міорелаксанти.
У разі супроводу аномалії Кіарі порушеннями неврологічних функцій або коли консервативний курс терапії не дає результатів, призначається хірургічна операція.
Часто при лікувальних курсахСиндром Кіарі використовується метод краніовертебральної декомпресії. Операція передбачає розширення отвору потиличної частини за рахунок видалення частини кісткової тканини, відсікання мозочкових мигдаликів та частини двох хребців шиї. Завдяки цьому нормалізується оборот цереброспінальної рідини в мозкових тканинах в результаті виконання латки з алотрансплантату або штучного матеріалу. Іноді синдром Кіарі лікують за допомогою шунтування, що дозволяє дренувати цереброспінальну рідину із центрального каналу. За допомогою хірургічної операціїможна відвести цереброспінальну рідину до судин органів грудей або очеревини.
Аномалія Кіар першого типу може протікати безсимптомно все життя. І третій тип патології майже завжди призводить до смертельного результату, якщо не здійснити своєчасне лікування. У разі появи неврологічних ознак захворювання першого або останнього типів дуже важливо своєчасно проведене хірургічне лікування, тому що недолік неврологічних функцій, що з'явився, буде погано відновлюватися, навіть якщо успішно провести маніпуляції. За різними даними, результативність хірургічної операції відзначається приблизно у половині епізодів.
Аномалію Арнольда-Кіарі відносять до вад краніо-вертебральної зони. Формується вона в задній черепній ямці (ЗЧЯ), при недостатньому обсязі якої відбувається зміщення задніх відділів мозку та мозочка в бік великого потиличного отвору, а також порушується струм спинномозкової рідини. Задня частина черепа утворює так звану задню черепну ямку, в якій розташовані півкулі та черв'як мозочка, міст, довгастий мозок, що переходить у спинний після проходження крізь великий потиличний отвір. Великий потиличний отвір обмежений кістковою основою черепа і не здатний змінювати діаметр, будь-які зміщення структур мозку загрожують невідповідністю їх розмірів і діаметра отвору, вклинюванням і утиском нервової тканини, наслідки якого можуть стати фатальними. У довгастому мозку сконцентровані життєво важливі нервові центри, які відповідають за діяльність серцево-судинної системита дихання, тому не тільки неврологічний дефіцит буде проявом захворювання. У важких випадках відбувається пригнічення вітальних функцій і хворий може померти. Зміщення гемісфер мозочка тягне зупинку циркуляції ліквору з гідроцефалією, яка ще більше посилює наявні розлади. Аномалія Арнольда-Кіарі буває вродженою, що формується у плода і поєднується з іншими відхиленнями у розвитку, а клініка її не завжди з'являється одразу. У частині випадків до маніфестації патології проходить значний проміжок часу або виникає ситуація, що провокує прояв безсимптомної раніше аномалії, в інших пацієнтів і зовсім вона може виявитися випадковою знахідкою на МРТ. Нерідко патологія має набутий характер і виникає під дією зовнішніх причинПри цьому мозок і череп при народженні мають нормальну будову.
Причини та механізм розвитку вади задньої черепної ямки (ЗЧЯ)
Єдиної думки щодо етіології аномалії Кіарі немає. Вчені висувають різні теорії, кожна з яких цілком обґрунтована та має право на існування.
Раніше аномалію вважали виключно вродженою порокомПроте спостереження фахівців показали, що лише мала частина пацієнтів мали дефекти під час внутрішньоутробного розвитку, інші ж їх набули вже в процесі життя.
Причинами набутої патології краніо-вертебрального переходу вважають нерівномірність швидкості росту нервової тканини мозку та кісткової основичерепа, коли мозок збільшується значно швидше, ніж кісткове вмістилище, в якому він знаходиться. Невідповідність обсягів, що виникає в кінцевому рахунку, і служить основою хвороби Арнольда-Кіарі. Вроджена формапатології поєднується з кістковою дисплазією, що тягне за собою недорозвинення кісток черепа, а також з порушенням формування зв'язкового апарату, і будь-яке зовнішній вплив, травма здатна різко посилити прояви патології Характерним вважається поєднання вади черепної ямки з іншими порушеннями утробного розвитку та уродженими синдромами. Неврологами сформульовано два основні механізми формування патології:
Оскільки аномалія може бути вродженою, серед причин вказують ті, які здатні змінити нормальний перебігвагітності та внутрішньоутробного розвитку:
Аномалія Арнольда-Кіарі виникає і з ряду набутих причин при правильно розвинених мозку і кістках черепа. Привести до її появи вже після народження здатні:
Гідроцефалія може бути провокуючим фактором, оскільки збільшення обсягу вмісту в черепі, нехай навіть за рахунок рідини, неминуче тягне за собою збільшення тиску і зміщення мозкових відділів у каутальному (задньому) напрямку. З іншого боку, вона є проявом самої аномалії, коли опущення мозочка викликає блокаду лікворних шляхів та наростання тиску спинномозкової рідини, що циркулює по порожнинах мозку.
Типи та ступеня аномалії Арнольда-Кіарі
Залежно від наявності тих чи інших змін з боку мозку та кісткової основи черепа, прийнято виділяти кілька різновидів аномалії Арнольда-Кіарі:
Аномалія Арнольда-Кіарі 1 типу - найчастіше діагностована і має досить сприятливий прогноз
аномалія Арнольда-Кіарі 3 типу на знімку
Що стосується степів тяжкості, то:
Аномалію Арнольда-Кіарі 1 ступеня можна вважати одним з найлегших варіантів патології, тому що пороків самого мозку при ній практично не виникає, а клініка може зовсім бути відсутнім, з'являючись лише за несприятливих умов - травма, нейроінфекція і т. д. Мальформації другий і третього ступеня, у свою чергу, часто поєднуються з різноманітними вадами розвитку нервової тканини - гіпоплазією деяких ділянок мозку та підкіркових вузлів, усуненням сірої речовини, кістами лікворних шляхів, недорозвиненням звивин мозку.
Прояви синдрому Арнольда-Кіарі
Симптоматика синдрому Арнольда-Кіарі визначається його типом та характером усунення структур ЗЧЯ. Часто він протікає безсимптомно і виявляється випадково під час обстежень мозку. У дорослих поява симптоматики може спровокувати травму голови, у малюків деякі форми захворювання помітні вже в перші години та дні життя. Аномалія I типу діагностується найчастіше і може проявитися у підлітковому чи дорослому віці наступними синдромами:
Гіпертензійний синдром викликаний збільшенням внутрішньочерепного тиску внаслідок блокади відтоку ліквору відділами мозку, що змістилися. Він проявляється:
Ознаками залучення мозочка (церебеллярний синдром) вважають розлади мови, рухової функції, рівноваги, ністагм. Пацієнти скаржаться на хиткість ходи, нестійкість положення тіла у просторі, утруднення дрібної моторикита чіткості рухів. Ураження стовбурового відділу мозку є небезпечним через розташування там ядер черепних нервів і життєво важливих нервових центрів. Стовбурова симптоматика полягає в:
Дорослі носії мальформації Арнольда-Кіарі вказують на наростання запаморочення та шуму у вухах, а також пароксизми втрати свідомості при повертанні та нахилах голови. Внаслідок здавлення стовбурів черепних нервів з'являється атрофія половини мови та порушення руху гортані з розладом акта ковтання, дихання та голосоутворення.
При утворенні порожнин і лікворних спинномозкових кіст на тлі утрудненого струму спинномозкової рідини у пацієнтів з I варіантом мальформації виникають ознаки сирингомієлічного синдрому – розлад чутливої сфери, оніміння шкіри, гіпотрофія м'язів, дисфункція тазових органів, зниження та зникнення черевних рефлексів, периферичні нейропатіїта зміни з боку суглобів. Розлади чутливості супроводжуються порушенням сприйняття власного тіла, коли пацієнт, заплющивши очі, не може сказати, в якому положенні знаходяться його руки чи ноги. Знижується також чутливість до болю та температури. За спостереженнями неврологів, діаметр і локалізація кісти спинного мозку не обов'язково позначаються на вираженості та поширеності порушень чутливої та рухової сфери, гіпотрофії м'язів. При синдромі другого і третього типу перебіг патології значно важчий, симптоматика з'являється у дитини відразу після пологів. Характерні порушення дихання – стридор (шумне дихання), напади його зупинки, а також двосторонній парез гортані, що провокує розлади ковтання, коли рідка їжа потрапляє у носові ходи. Другий тип аномалії у малюків перших місяців життя супроводжується ністагмом, посиленням тонусу м'язів у руках, синюшністю шкіри, які особливо помітні при годівлі немовляти. Рухові порушенняваріабельні, прояви їх змінюються, можлива тетраплегія – параліч і верхніх, і нижніх кінцівок.
Аномалія Арнольда-Кіарі третього та четвертого варіантів протікає важко,це вроджена патологія, яка сумісна з нормальної життєдіяльністю, тому прогноз за такого діагнозу не можна вважати сприятливим.
Мальформація Арнольда-Кіарі може призводити до ускладнень, викликаних блокадою струму ліквору, ураженням ядер черепних нервів, утиском стовбурових структур. Найчастішими вважаються:
При тяжкому перебігупатології може наступити кома, зупинка серця та дихання, які призводять до смерті за лічені хвилини. Реанімаційні заходидозволяють забезпечити вітальні функції, але повернути до життя мозок та усунути незворотні наслідки компресії його відділів, на жаль, практично неможливо.
Діагностика та лікування мальформації Арнольда-Кіарі
За особливостями симптоматики та на підставі огляду невролога діагноз мальформації Кіарі поставити неможливо. Енцефалографія, дослідження судин голови теж не дадуть жодної інформації щодо причин неврологічних порушень, проте можуть показати наявність підвищеного тискуу черепі. Рентгенографія, КТ, МСКТ вкажуть на наявність дефектів кісток черепа, які характерні для цієї патології, але стан м'якотканих структур, нервової тканини встановити при цьому не можна. Точна діагностика аномалії стала можлива завдяки використанню МРТ,за допомогою якої лікар може визначити і кісткові вади, і варіанти розвитку самого мозку, його судин, рівень розташування відділів щодо черепних кісток, їх розміри, об'єм задньої черепної ямки та ширину великого потиличного отвору. МРТ можна вважати єдиним точним і достовірним методом виявлення патології. МРТ вимагає знерухомлення пацієнта, який має якийсь час спокійно лежати на столі апарата. У дітей із цим можуть виникнути значні труднощі, тому дослідження проводять у стані медикаментозного сну. Для пошуку поєднаних вад спинного мозку та хребта досліджують також ці відділи хребетного стовпа.
Коли встановлено діагноз, пацієнта направляють до нейрохірурга або невролога для визначення плану лікування, показань до хірургічної операції, її виду.
Аномалія Арнольда-Кіарі, яка протікає безсимптомно, не потребує лікування. Більше того, і сам носій патології може не здогадуватися, що в організмі щось не так. З появою клінічних ознакзахворювання показано консервативне чи оперативне лікування. Якщо прояви обмежені головним болем, призначається медикаментозна терапія , що включає протизапальні засоби (найз, ібупрофен, диклофенак), анальгетики (кеторол) та препарати, що знімають спазм м'язів (мідокалм). За наявності неврологічних розладів, ознак здавлення відділів мозку, нервових стовбурів, у разі відсутності ефекту від медикаментозного лікуванняпротягом 3 місяців, хворому необхідна хірургічна корекція.Операція необхідна для усунення здавлювання нервової тканини та нормалізації циркуляції ліквору. Найпопулярнішою операцією при захворюванні Кіарі вважається краніовертебральна декомпресія, спрямована на збільшення розмірів ЗЧЯ. При декомпресії хірург видаляє ділянки потиличної кістки, резецирует мигдалики мозочка, за потребою проводить висічення задніх відділів перших шийних хребців. Для попередження пролабування задніх частинмозку в отриманий отвір на тверду мозкову оболонку накладаються спеціальні синтетичні латки.
Приклад видалення частин потилицею кістки та шийних хребців Декомпресія ЗЧЯ вважається і травматичною, і ризикованою. Статистика така, що ускладнення виникають щонайменше у кожного десятого хворого, тоді як без операції показник смертності набагато нижчий. У зв'язку з високим ризикомоперативного лікування нейрохірурги вдаються до нього лише у разі справді серйозних показань – клініки здавлення ділянок мозку. Іншим варіантом хірургічного лікування вважається шунтування, яке забезпечує відтік ліквору з порожнини черепа до грудної або черевної порожнини. Шляхом імплантації спеціальних трубок відбувається перетік спинномозкової рідини та зниження внутрішньочерепного тиску. При тяжких формах патології показано госпіталізація, заходи щодо профілактики інфекційних ускладненьта корекції неврологічних розладів. Наростання набряку мозку внаслідок вклинювання його відділів у потиличний отвір вимагають лікування в умовах реанімації, що включає боротьбу з набряком (магнезія, фуросемід, діакарб), налагодження штучної вентиляціїлегень у разі порушення дихання тощо.
Тривалість життя та прогноз при аномалії Арнольда-Кіарі залежать від типу патології. При I типі прогноз вважатимуться сприятливим, часом клініка немає або провокується лише сильними травмуючими чинниками. При безсимптомному перебігуносії аномалії живуть стільки ж, скільки й інші люди. При аномалії другого та першого типу з клінічними проявамипрогноз дещо гірший, тому що проявляється неврологічний дефіцит, який складно усунути навіть при активному лікуванніТому для таких пацієнтів велике значення має своєчасно проведена операція. Чим раніше хворому буде надано хірургічна допомога, Тим не менш виражені неврологічні зміни на нього чекають. Мальформація третього і четвертого типів - найбільш важкі формипатології. Прогноз несприятливий, тому що залучаються багато структур мозку, часто мають місце поєднані вади інших органів, тяжкі порушення функції стовбура мозку, не сумісні з життям.
Відео: презентація та ін. інформація з аномалії Арнольда-Кіарі
Крок 1: сплатіть консультацію за допомогою форми → Крок 2: після оплати задайте своє запитання у форму нижче ↓ Крок 3:Ви можете додатково віддячити фахівцю ще одним платежем на довільну суму

У 2018 році Різдвяний пост розпочнеться 28 листопада. У цей період православні віруючі готуються до зустрічі Різдва.

Створення сім'ї – мрія більшості жінок. Вони хочуть мати люблячого чоловіка і купу дітей. Тільки от не завжди стосунки...

Ця стаття містить: найсильніша молитва від розлучення - інформація взята зі всіх куточків світла, електронної мережі та...

Інформаційний сайт про ікони, молитви, православні традиції. Молитва від скандалів та сварок у сім'ї, з чоловіком, з дітьми...

Який же Новий рік без шампанського, мандарин, «Олів'є», заливного та всіма улюбленої «Оселедця під шубою». Ось із останнім...

Підготуємо необхідні інгредієнти для печива. Перше, що потрібно зробити - це поставити воду кип'ятитися. Нам необхідний...

Чи можна оформити працівника посаду фінансовий директор - головний бухгалтер. Головний бухгалтер стверджує, що...

Керівник невеликого бізнесу може вести бюджет самостійно. ПЕРЕВІРЕНО! Якщо займатися веденням...

Створення нових проектів передбачає попереднє економічне обґрунтування їхньої доцільності, наступне...
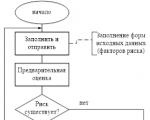
Звітність формується РМ, проходить узгодження (схвалення) Комітету з ризиків за Правління та передається на...

На краю великого луга, на довгій смарагдовій травинці жила крихітна Божа Корівка. Маленька Божа...

Нині досить поширене звернення людини до зірок. За допомогою гороскопу людина може дізнатися...

Діловий чи приятельський. Якщо ж переслідували незнайомку - значить рівень вашої довіри до жіночої статі.

по соннику ФрейдаЯкщо вам снилося, як ви ловили рибу, значить, у реальному житті ви насилу можете відключитися...

Створення сім'ї – мрія більшості жінок. Вони хочуть мати люблячого чоловіка і купу дітей. Тільки от не завжди...

Ця стаття містить: найсильніша молитва від розлучення - інформація взята зі всіх куточків світла, електронної...