Herring under a fur coat - a classic recipe
What would New Year be without champagne, tangerines, Olivier, aspic and everyone’s favorite “Herring under a fur coat”. With the last one...
Dystrophy (from the Greek dys - disorder and tropho - nourish) is quantitative and qualitative structural changes in the cells and/or intercellular substance of organs and tissues caused by a disorder metabolic processes. In dystrophies, as a result of trophic disturbances, they accumulate in cells or in the intercellular substance. various products metabolism (proteins, fats, carbohydrates, minerals, water). The morphological essence of dystrophies is expressed in:
Thus, dystrophy is a morphological expression of metabolic disorders of cells and tissues.
Among the mechanisms for maintaining normal trophism, cellular and extracellular are distinguished.
Cellular mechanisms are provided by the structural organization of the cell and its autoregulation, provided by the genetic code.
Extracellular mechanisms of trophism are provided by transport (blood, lymph) and integrative (nervous, endocrine, humoral) systems of its regulation.
The direct cause of the development of dystrophies can be:
ü Hypoglycemia. Low level Blood glucose (hypoglycemia) leads to insufficient production of adenosine triphosphate (ATP) molecules, which is most pronounced in the brain.
ü Hypoxia: Lack of oxygen in cells (hypoxia) can occur when:
Morphogenesis of dystrophies
Among the mechanisms leading to the development of changes characteristic of dystrophies, there are infiltration, decomposition (phanerosis), perverted synthesis and transformation.
Infiltration - excessive penetration of metabolic products from the blood and lymph into cells or intercellular substance and/or disruption of their inclusion in metabolism with subsequent accumulation. For example, infiltration of the epithelial protein of the proximal tubules of the kidneys in nephrotic syndrome, infiltration of the intima of the aorta and large arteries with lipoproteins in atherosclerosis.
Decomposition (phanerosis) - decomposition of chemically complex substances. For example, the breakdown of lipoprotein complexes and the accumulation of free fat in the cell ( fatty degeneration cardiomyocytes during diphtheria intoxication). The breakdown of polysaccharide-protein complexes underlies fibrinoid changes in connective tissue in rheumatic diseases.
Transformation - the transition of one substance to another. This is, for example, the transformation of carbohydrates into fats during diabetes mellitus, enhanced polymerization of glucose into glycogen, etc.
Perverted Synthesis - is the synthesis in cells or tissues of substances that are not normally found in them. These include: the synthesis of abnormal amyloid protein in the cell and the formation of abnormal amyloid protein-polysaccharide complexes in the intercellular substance, the synthesis of alcoholic hyaline protein by hepatocytes, the synthesis of glycogen in the epithelium of a narrow segment of the nephron in diabetes mellitus.
Classification.
Depending on the location of metabolic disorders:
ü parenchymal;
ü stromal-vascular;
ü mixed.
According to the predominance of violations of one or another type of exchange:
ü protein;
ü fatty;
ü carbohydrates;
ü mineral.
Depending on the influence of genetic factors:
ü purchased;
ü hereditary.
According to the prevalence of the process:
ü local.
PARANCHYMATOUS DYSTROPHY
Since the time of R. Virchow, many pathologists have and continue to classify the so-called “granular dystrophy”, which R. Virchow himself designated as “cloudy swelling,” among parenchymal protein dystrophies. This is how it is customary to designate the process in which pronounced granularity appears in the cytoplasm of the cells of parenchymal organs. The organs increase in size, become flabby and dull when cut, as if scalded by boiling water. Electron microscopic and histoenzyme-chemical studies of “granular dystrophy” showed that it is based not on the accumulation of protein in the cytoplasm, but on hyperplasia (i.e., an increase in the number) of ultrastructures of cells of parenchymal organs as an expression of the functional tension of these organs in response to various influences.
PARANCHYMATOUS PROTEIN DYSTROPHIES
Currently, parenchymal protein dystrophies (dysproteinoses) include hyaline-droplet, hydropic and horny. However, it should be emphasized that horny dystrophy, according to the mechanism of its development, is not related to the previous ones.
Hyaline droplet dystrophy
With hyaline-droplet dystrophy, large hyaline-like protein lumps and droplets appear in the cytoplasm, merging with each other and filling the cell body. The basis of this dystrophy is the coagulation of cytoplasmic proteins with pronounced destruction of the ultrastructural elements of the cell - focal coagulative necrosis.
This type of dysproteinosis often occurs in the kidneys, less often in the liver, and very rarely in the myocardium.
The appearance of organs with this dystrophy does not have any characteristic features. Macroscopic changes are characteristic of those diseases in which hyaline-droplet dystrophy occurs.
In the kidneys, upon microscopic examination, the accumulation of large grains of bright pink protein - hyaline drops - is found in nephrocytes. In this case, destruction of mitochondria, endoplasmic reticulum, and brush border is observed. The basis of hyaline-droplet dystrophy of nephrocytes is the insufficiency of the vacuolar-lysosomal apparatus of the epithelium of the proximal and distal convoluted tubules, which normally reabsorbs proteins. Therefore, this type of nephrocyte dystrophy is very common in nephrotic syndrome and reflects the reabsorption insufficiency of the convoluted tubules in relation to proteins. This syndrome is one of the manifestations of many kidney diseases in which the glomerular filter is primarily affected (glomerulonephritis, renal amyloidosis, paraproteinemic nephropathy, etc.)
In the liver, upon microscopic examination, lumps and drops of a protein nature are found in hepatocytes - this is alcoholic hyaline, which at the ultrastructural level represents irregular aggregates of microfibrils and irregularly shaped hyaline inclusions (Mallory bodies). The formation of this protein and Mallory bodies is a manifestation of the perverted protein-synthetic function of the hepatocyte and is constantly detected in alcoholic hepatitis.
The outcome of hyaline droplet dystrophy is unfavorable: it ends in an irreversible process leading to total coagulative necrosis of the cell.
The functional significance of this dystrophy is very great - it occurs a sharp decline organ functions. Hyaline-droplet dystrophy of the epithelium of the renal tubules is associated with the appearance of protein (proteinuria) and casts (cylindruria) in the urine, loss of plasma proteins (hypoproteinemia), and disruption of electrolyte balance. Hyaline droplet degeneration of hepatocytes is often the morphological basis of disorders of many liver functions.
Hydropic dystrophy
It is characterized by the appearance in the cell of vacuoles filled with cytoplasmic fluid. The fluid accumulates in the cisterns of the endoplasmic reticulum and in the mitochondria, less often in the cell nucleus. The mechanism of development of hydropic dystrophy is complex and reflects disturbances in water-electrolyte and protein metabolism, leading to changes in colloid osmotic pressure in the cell. A major role is played by disruption of the permeability of cell membranes, accompanied by their disintegration. This leads to the activation of lysosome hydrolytic enzymes, which break intramolecular bonds with the addition of water. Essentially, such cell changes are an expression of focal liquefaction necrosis.
Hydropic dystrophy is observed in the epithelium of the skin and renal tubules, in hepatocytes, muscle and nerve cells, as well as in the cells of the adrenal cortex.
The reasons for the development of hydropic dystrophy in different organs are ambiguous. In the kidneys, this is damage to the glomerular filter (glomerulonephritis, amyloidosis, diabetes mellitus), which leads to hyperfiltration and insufficiency of the nephrocyte enzyme system, which normally ensures water reabsorption; glycol poisoning, hypokalemia. In the liver, hydropic dystrophy occurs with viral and toxic hepatitis. The causes of hydropic dystrophy of the epidermis can be infections and allergies.
The appearance of organs and tissues changes little with hydropic dystrophy.
Microscopic picture: parenchymal cells are increased in volume, their cytoplasm is filled with vacuoles containing clear liquid. The nucleus shifts to the periphery, sometimes vacuolates or shrinks. An increase in hydropia leads to the disintegration of cell ultrastructures and overflow of the cell with water, the appearance of liquid-filled balloons, therefore such changes are called balloon dystrophy.
The outcome of hydropic dystrophy is usually unfavorable; it ends with total colliquation necrosis of the cell. Therefore, the function of organs and tissues in hydropic dystrophy is sharply reduced.
Horny dystrophy
Horny dystrophy, or pathological keratinization, is characterized by excessive formation of horny substance in the keratinizing epithelium (hyperkeratosis, ichthyosis) or the formation of horny substance where it normally does not exist - pathological keratinization on the mucous membranes, for example, in the oral cavity (leukoplakia), esophagus, cervix. Horny dystrophy can be local or general, congenital or acquired.
The causes of horny dystrophy are varied: chronic inflammation associated with infectious agents, the action of physical and chemical factors, vitamin deficiencies, congenital disorders of skin development, etc.
The outcome can be twofold: elimination causing cause at the beginning of the process it can lead to tissue restoration, but in advanced cases cell death occurs.
The significance of horny dystrophy is determined by its degree, prevalence and duration. Long-term pathological keratinization of the mucous membrane (leukoplakia) can be a source of development cancerous tumor. Severe congenital ichthyosis is, as a rule, incompatible with life.
PARANCHYMATOUS FATTY DYSTROPHY (LIPIDOSES)
Parenchymal fatty degeneration is a structural manifestation of a disorder in the metabolism of cytoplasmic lipids, which can be expressed in the accumulation of fat in a free state in cells where it is not found normally.
The causes of fatty degeneration are varied:
Parenchymal fatty degeneration is characterized mainly by the accumulation of triglycerides in the cytoplasm of parenchymal cells. When the connection of proteins with lipids is disrupted - decomposition, which occurs under the influence of infections, intoxications, lipid peroxidation products - destruction of the membrane structures of the cell occurs and free lipoids appear in the cytoplasm, which are the morphological substrate of parenchymal fatty degeneration. It is most often observed in the liver, less often in the kidney and myocardium, and is regarded as a nonspecific response to a large number of types of damage.
Microscopic signs of fatty degeneration: any fat found in tissues dissolves in solvents that are used to stain tissue samples for microscopic examination. Therefore, with conventional wiring and tissue staining (hematoxylin and eosin staining), cells in the most early stages fatty degeneration have pale and foamy cytoplasm. As fatty inclusions increase, small vacuoles appear in the cytoplasm.
Fat-specific staining requires the use of frozen sections made from fresh tissue. In frozen sections, the fat remains in the cytoplasm, after which the sections are stained with special dyes. Histochemically, fats are detected using a number of methods: Sudan IV, fatty red O and scarlet mouth stain them red, Sudan III - orange, Sudan black B and osmic acid - black, Nile blue sulfate stains fatty acids dark blue. , and neutral fats - in red. Using a polarizing microscope, it is possible to differentiate between isotropic and anisotropic lipids. Anisotropic lipids such as cholesterol and its esters exhibit characteristic birefringence.
Fatty liver degeneration is manifested by a sharp increase in the content and change in the composition of fats in hepatocytes. In the liver cells, lipid granules first appear (pulverized obesity), then small droplets of them (small-droplet obesity), which subsequently merge into large droplets (large-droplet obesity) or into one fat vacuole, which fills the entire cytoplasm and pushes the nucleus to the periphery. Liver cells modified in this way resemble fat cells. More often, fat deposition in the liver begins at the periphery, less often - in the center of the lobules; with significantly pronounced dystrophy, liver cell obesity is diffuse.
Macroscopically, the liver in fatty degeneration is enlarged, anemic, doughy in consistency, has a yellow or ocher-yellow color, with greasy shine on the cut. When making a cut, a coating of fat is visible on the knife blade and the cut surface.
Types of fatty liver:
Acute fatty liver is a rare but serious condition associated with acute lesion liver. In acute fatty liver, triglycerides accumulate in the cytoplasm as small, membrane-bounded vacuoles (small fatty liver).
Chronic fatty liver can occur with chronic alcoholism, malnutrition and poisoning with certain hepatotoxins. Fat droplets in the cytoplasm combine to form large vacuoles (large droplet fatty liver disease). The localization of fatty changes in the liver lobule depends on the reasons that caused them. Even with severe chronic fatty liver, there are rarely clinical manifestations liver dysfunction.
Myocardial fatty degeneration is characterized by the accumulation of triglycerides in the myocardium.
Causes of fatty degeneration of the myocardium:
chronic hypoxic conditions, especially with severe anemia. In chronic fatty degeneration, yellow stripes alternate with red-brown areas (“tiger heart”). Clinical signs usually not very pronounced.
toxic damage, such as diphtheritic myocarditis, causes acute fatty degeneration. Macroscopically, the heart is flabby, there is a yellow diffuse staining, the heart looks enlarged in volume, its chambers are stretched; V clinical picture signs of acute heart failure appear.
Fatty degeneration of the myocardium is considered as the morphological equivalent of its decompensation. Most of the mitochondria disintegrate, and the cross-striations of the fibers disappear. The development of fatty degeneration of the myocardium is most often associated not with the destruction of complexes cell membranes, and with the destruction of mitochondria, which leads to impaired oxidation fatty acids in a cage. In the myocardium, fatty degeneration is characterized by the appearance of tiny fat droplets in muscle cells (pulverized obesity). With increasing changes, these droplets (small-droplet obesity) completely replace the cytoplasm. The process is focal in nature and is observed in groups of muscle cells located along the venous knee of capillaries and small veins, most often subendo- and subepicardial.
In the kidneys, with fatty degeneration, fats appear in the epithelium of the proximal and distal tubules. Usually these are neutral fats, phospholipids or cholesterol, which are found not only in the tubular epithelium, but also in the stroma. Neutral fats in the epithelium of the narrow segment and collecting ducts occur as a physiological phenomenon.
Appearance of the kidneys: they are enlarged, flabby (dense when combined with amyloidosis), the cortex is swollen, gray with yellow specks, noticeable on the surface and section.
The mechanism of development of fatty kidney degeneration is associated with infiltration of the epithelium of the renal tubules with fat during lipemia and hypercholesterolemia (nephrotic syndrome), which leads to the death of nephrocytes.
The outcome of fatty degeneration depends on its degree. If it is not accompanied by gross breakdown of cellular structures, then, as a rule, it turns out to be reversible. A profound disruption of cellular lipid metabolism in most cases ends in cell death.
The functional significance of fatty degeneration is great: the functioning of organs is sharply disrupted, and in some cases stops. Some authors have expressed the idea that fat appears in cells during the period of convalescence and the beginning of repair. This is consistent with biochemical ideas about the role of the pentose phosphate pathway for glucose utilization in anabolic processes, which is also accompanied by the synthesis of fats.
PARANCHYMATOUS CARBOHYDRATE DYSTROPHIES
Carbohydrates, which are determined in cells and tissues and can be identified histochemically, are divided into polysaccharides, of which only glycogen, glycosaminoglycans (mucopolysaccharides) and glycoproteins are detected in animal tissues. Among glycosaminoglycans, there are neutral ones, tightly bound to proteins, and acidic ones, which include hyaluronic acid, chondroitinsulfuric acid and heparin. Acidic glycosaminoglycans, as biopolymers, are capable of forming weak compounds with a number of metabolites and transporting them. The main representatives of glycoproteins are mucins and mucoids. Mucins form the basis of mucus produced by the epithelium of the mucous membranes and glands; mucoids are part of many tissues.
Histochemical methods for identifying carbohydrates. Polysaccharides, glycosaminoglycans and glycoproteins are detected by the PAS reaction. The essence of the reaction is that after oxidation with periodic acid (or reaction with potassium periodate), the resulting aldehydes give a red color with Schiff fuchsin. To detect glycogen, the PHIK reaction is supplemented with enzymatic control - treatment of sections with amylase. Glycogen is stained red by Best's carmine. Glycosaminoglycans and glycoproteins are determined using a number of methods, of which the most commonly used are toluidine blue or methylene blue stains. These stains make it possible to identify chromotropic substances that give rise to the metachromasia reaction. Treatment of tissue sections with hyaluronidase (bacterial, testicular) followed by staining with the same dyes makes it possible to differentiate different glycosaminoglycans; this is also possible by changing the pH of the dye.
Parenchymal carbohydrate dystrophy may be associated with impaired glycogen or glycoprotein metabolism.
Glycogen metabolism disorder
In diabetes mellitus, the development of which is associated with pathology of the β-cells of the pancreatic islets, which causes insufficient insulin production, there is insufficient use of glucose by tissues, an increase in its content in the blood (hyperglycemia) and excretion in the urine (glucosuria). Tissue glycogen reserves decrease sharply. This primarily concerns the liver, in which glycogen synthesis is disrupted, which leads to its infiltration with fats - fatty liver degeneration develops; at the same time, inclusions of glycogen appear in the nuclei of hepatocytes, they become light ("empty" nuclei).
Associated with glucosuria characteristic changes kidneys in diabetes. They are expressed in glycogen infiltration of the tubular epithelium, mainly in the narrow and distal segments. The epithelium becomes tall, with light foamy cytoplasm; glycogen grains are also visible in the lumen of the tubules. These changes reflect the state of glycogen synthesis (glucose polymerization) in the tubular epithelium during the resorption of glucose-rich plasma ultrafiltrate.
In diabetes, not only the renal tubules are affected, but also the glomeruli and their capillary loops, the basement membrane of which becomes permeable to sugars and plasma proteins. One of the manifestations of diabetic microangiopathy occurs - intercapillary (diabetic) glomerulosclerosis.
Hereditary carbohydrate dystrophies, which are based on disorders of glycogen metabolism, are called glycogenoses. Glycogenosis is caused by the absence or deficiency of the enzyme involved in the breakdown of stored glycogen, and therefore belongs to hereditary enzymopathies, or storage diseases. Currently, 6 types of glycogenosis caused by hereditary deficiency 6 different enzymes. These are Gierke (type I), Pompe (type II), McArdle (type V) and Hers (type VI) diseases, in which the structure of glycogen accumulated in the tissues is not disturbed, and Forbes-Cory (type III) and Andersen diseases ( IV type), in which it is sharply changed.
Morphological diagnosis of glycogenosis of one type or another is possible by examining a biopsy using histoenzyme methods, as well as taking into account the localization of accumulated glycogen.
Carbohydrate dystrophies associated with impaired glycoprotein metabolism
When the metabolism of glycoproteins in cells or in the intercellular substance is disrupted, mucins and mucoids, also called mucous or mucus-like substances, accumulate. In this regard, when glycoprotein metabolism is disrupted, they speak of mucous dystrophy.
Microscopic examination. It makes it possible to detect not only increased mucus formation, but also changes in the physicochemical properties of mucus. Many secreting cells die and desquamate, the excretory ducts of the glands become obstructed by mucus, which leads to the development of cysts. Often in these cases inflammation is associated. Mucus can close the lumens of the bronchi, resulting in the occurrence of atelectasis and foci of pneumonia.
Sometimes it is not true mucus that accumulates in the glandular structures, but mucus-like substances (pseudomucins). These substances can become denser and take on the character of a colloid. Then they talk about colloid dystrophy, which is observed, for example, with colloid goiter.
The causes of mucous dystrophy are varied, but most often it is inflammation of the mucous membranes as a result of the action of various pathogenic irritants (catarrhal inflammation).
Mucosal dystrophy underlies hereditary systemic disease, called cystic fibrosis, which is characterized by a change in the quality of mucus secreted by the epithelium of the mucous glands: the mucus becomes thick and viscous, it is poorly excreted, which causes the development of retention cysts and sclerosis ( cystic fibrosis). The exocrine apparatus of the pancreas, glands of the bronchial tree, digestive and urinary tracts are affected, biliary tract, sweat and lacrimal glands.
The outcome is largely determined by the degree and duration of excess mucus production. In some cases, regeneration of the epithelium leads to full restoration mucous membrane, in others it atrophies and subsequently becomes sclerotic, which naturally affects the function of the organ.
Dystrophy of cells and tissues is a violation of tissue or cellular metabolism, accompanied by certain structural changes in cells and intercellular substance.The development of dystrophy is based on disorders of the regulatory mechanisms of trophism of a congenital or acquired nature (hereditary and acquired dystrophy of cells and tissues).
Depending on the predominance of morphological changes in the parenchyma cells or stroma of organs, dystrophies are divided into parenchymal, mesenchymal and mixed. The predominance of disturbances of one or another type of metabolism underlies the release of protein, fat, carbohydrate and mineral dystrophies, and the prevalence of the process determines their division into general (systemic) and local.
Morphogenetic mechanisms of dystrophy include infiltration - deposition of coarsely dispersed proteins or lipids in cells or extracellular substances; synthesis of abnormal substances (for example amyloid); transformation (for example, carbohydrates and proteins into fats) and decomposition (phanerosis) - the breakdown of lipoproteins in the membrane structures of the cell with the release of lipids and proteins.
Mesenchymal lipidoses occur when there is a disturbance in the metabolism of neutral fat or cholesterol and its esters; they can be general or local. An increase in neutral fat in fat depots is called general obesity, a decrease is called exhaustion. A local decrease in the amount of adipose tissue is characteristic of regional lipodystrophy; its local increase is possible with tissue or organ atrophy (fat replacement), with some endocrine disorders. Impaired cholesterol metabolism is most clearly manifested in atherosclerosis.
With a hereditary deficiency of enzymes that metabolize certain types of lipids, systemic lipidoses (hereditary enzymopathies) occur: cerebrosidosis (Gaucher disease), sphingomyelinosis (Niemann-Piquet disease), gangliosidosis (Tay-Sachs disease, or amaurotic idiocy), generalized gangliosidosis, etc.
Disorders of glycogen metabolism are manifested by a decrease or increase in its content in tissues, appearing where it is usually not found. In diabetes mellitus, tissue glycogen reserves sharply decrease and its synthesis is disrupted. As a result of glycosuria, glycogen infiltration of the epithelium of the renal tubules occurs, and glycogen grains appear in their lumens. The glomeruli are also affected. With glycogenosis, glycogen accumulates in the liver, kidneys, skeletal muscles, myocardium, spleen.
Mesenchymal carbohydrate dystrophies are manifested by sliming of the main substance (mucosal dystrophy) of the connective tissue and are associated with impaired metabolism of glycoproteins and mucopolysaccharides (glycosaminoglycans). The cause of such dystrophies most often lies in dysfunction endocrine glands or exhaustion (for example, mucous swelling or myxedema with hypofunction of the thyroid gland, mucus of connective tissue with cachexia).
Definition
Granular dystrophy is a parenchymal dystrophy, characterized by the appearance in the cytoplasm of granule cells, which are swollen mitochondria. Considered as a type of protein dystrophy.
Occurrence.
Most often observed in hepatocytes, nephrocytes, cardiomyocytes. The phenomenon is extremely common, since it manifests itself mainly during circulatory hypoxia, which is observed in many pathological conditions.
Conditions of occurrence.
Mechanisms of occurrence.
Under hypoxic conditions, oxidative phosphorylation and ATP synthesis sharply decrease. Due to energy deficiency, the work of the ion pump - K + /Na + - ATPase - built into the membranes of organelles and the cell membrane and ensuring the active removal of Na + ions outside the cell is disrupted. In organelles, primarily in mitochondria, accumulation of these ions and, consequently, water occurs. Swollen mitochondria take on the appearance of grains, visible under a light microscope.
Macroscopic picture.
Change internal organs with this type of dystrophy it is described as “cloudy swelling”. In the kidneys and liver, which have a capsule, the tissue bulges somewhat beyond the edges of the cut. The bulging is associated with the same dysfunction of ion pumps and with the accumulation of excess amounts of water in the cell. The cloudy sheen and dull appearance of an organ on a section is probably due to the fact that the surface layer of the section turns out to be optically denser due to swollen mitochondria and reflects worse the light falling on it, which we perceive.
Microscopic picture.
In the cytoplasm of cells, at high magnification, small grains with a saturated pink color when stained with eosin. They are especially noticeable when the microspecimen is stained with azure and eosin. At the ultrastructural level, mitochondria are enlarged by 2-5 times or more with an enlightened matrix, with fragmented, often poorly visible cristae. The outer layer of the bilayer membrane is absent in a number of organelles. In some cases, changes in mitochondria are accompanied by expansion of the cisterns of the rough endoplasmic reticulum.
Clinical significance.
The very fact of granular dystrophy indicates an oxygen and energy deficiency of the cell, which has an extremely unfavorable effect on its function, especially when it comes to cardiomyocytes. On the other hand, swelling of mitochondria and fragmentation of cristae in them further disrupts ATP synthesis. The normally existing “dance of mitochondria” is also sharply disrupted: mitochondria that have become “clumsy” poorly ensure the delivery of ATP to energy-consuming organelles, which cannot but adversely affect the functioning of the entire cell.
Granular dystrophy can occur within a few minutes, also quickly disappearing when the normal oxygen regime in the cell is restored: ion pumps pump out both excess Na 5 + and water from the mitochondria, the mitochondria swell, and those whose outer layer of the membrane is destroyed, are captured by lysosomes and utilized.
Lecture outline:
The concept of alteration.
Dystrophies like pathological process. Mechanisms. Classification.
Parenchymal dystrophies.
Mesenchymal dystrophies.
Mixed dystrophies.
Disorders of mineral metabolism.
Necrosis: causes, signs.
Atrophy: causes, types.
Damage, or alteration, is called a change in cells, intercellular substance, and depending on the volume of damaged cells - tissues and organs. IN damaged cells, tissues and organs, metabolism changes, which leads to disruption of their vital functions and usually to dysfunction. Damage accompanies any disease or pathological process. At the same time, the damage itself causes the formation of substances that promote the activation of protective and regenerative reactions. If these reactions are sufficient to eliminate the damage, recovery occurs. In cases where protective-adaptive reactions are insufficient, damage becomes irreversible and tissue death develops with a decrease or complete loss of organ functions. Finally, in cases where the volume and severity of damage increases and is not compensated by the body’s adaptive reactions, the patient’s death occurs.
Among the damages, the most important ones are dystrophy, necrosis and atrophy. The expression of the most profound and irreversible changes that occur in the body due to various injuries is death.
Dystrophy– a pathological process reflecting metabolic disorders in the body. Dystrophy is characterized by damage to cells and intercellular substance, as a result of which the function of the organ changes.
Dystrophy is based on a violation of trophism, i.e., a set of mechanisms that ensure metabolism and the preservation of the structure of cells and tissues.
Cellular mechanisms are provided by the very structure of the cell and its self-regulation, due to which each cell performs its characteristic function.
Extracellular mechanisms include a system for transporting metabolic products (blood and lymphatic microvasculature), a system of intercellular structures of mesenchymal origin and a system of neuroendocrine regulation of metabolism. If there is a violation in any link of the trophic mechanisms, one or another type of dystrophy may occur.
The essence of dystrophy lies in the fact that in cells or the intercellular space an excess or insufficient amount of compounds characteristic of them is formed, or substances are formed that are not characteristic of a given cell or tissue. There are several mechanisms for the development of dystrophy.
MECHANISMS OF DEVELOPMENT OF DYSTROPHIES
Infiltration, in which substances characteristic of it enter the cell with the blood, but in greater quantities than normal. For example, infiltration of cholesterol and its derivatives into the intima of large arteries in atherosclerosis.
Perverted synthesis in which abnormal, i.e., are formed in cells or intercellular substance. substances not characteristic of these cells and tissues. For example, under certain conditions, cells synthesize amyloid protein, which is not normally present in humans.
Transformation, in which, for certain reasons, instead of products of one type of metabolism, substances characteristic of another type of metabolism are formed, for example, proteins are transformed into fats or carbohydrates .
Decomposition, or phanerosis. With this mechanism, dystrophy develops as a result of the breakdown of complex chemical compounds that make up cellular or intercellular structures. For example, the disintegration of membranes of intracellular structures consisting of fat-protein complexes during hypoxia leads to the appearance in the cell of an excess amount of either proteins or fats. Protein or fatty degeneration occurs.
Depending on the degree of metabolic disorder and the severity of morphological changes, dystrophies can be reversible or irreversible. In the latter case, the pathological process will progress until the death (necrosis) of the cell or tissue. Consequently, the outcome of irreversible dystrophies is necrosis.
Alteration(Latin alteratio – change) or damage are structural changes in the cells and tissues of the body that occur under the influence of exogenous and endogenous factors. At the same time, metabolism, function and vital activity in tissues and organs are disrupted. The causes of alteration may be circulatory disorders, physical agents and chemical substances, infectious agents, immunopathological reactions, genetic factors and an imbalance of substances necessary for the cell (usually due to nutritional disorders). The degree of damage to cells and tissues depends on the type and duration of action of the pathogenic factor, on the morpho-functional characteristics of the macroorganism. Alteration can occur at the ultrastructural, cellular, tissue and organ levels.
In the pathogenesis of cell and tissue damage great importance play hypoxic factors associated with insufficient oxygen supply to tissues (for example, when the flow of arterial blood is obstructed). For hypoxia
Oxidative phosphorelation stops and ATP synthesis decreases, glycolysis is activated. Acute swelling (edema) of the cell occurs due to a violation of osmotic pressure and accumulation of metabolites (phosphates, lactate, etc.) in the cytoplasm, the cisterns of the endoplasmic reticulum swell, mitochondria expand, plasma membranes are damaged, including lysosome membranes, which leads to the release their enzymes and the breakdown of cell components.
Cell damage can be caused by oxygen free radicals, chemical factors, and ionizing radiation; develops during cell aging. In this case, peroxidation of membrane lipids occurs, which can lead to the destruction of the plasma membrane and organelles. A significant role is also played by the oxidative transformation of proteins, which enhances the destruction of key enzymes through neutral proteases, and damage to cell DNA by free radicals.
Damage can be caused by chemicals that can act directly on cell molecules and organelles (for example, water-soluble mercuric chloride compounds).
Alteration is represented by two pathological processes - dystrophy and necrosis, which can be successive stages.
Dystrophy(Greek dys – prefix denoting disorder and trophe – nutrition) – a complex pathological process, which is based on metabolic disorders leading to structural changes. In morphological terms, during degeneration of cells and tissues, substances appear that are normally absent or contained in small quantities, or substances inherent to them disappear from cells and tissues.
Dystrophy is based on a violation of a complex of mechanisms that ensure metabolism and the preservation of cell and tissue structures, such as the regulatory function of the nervous and endocrine systems, disorders of blood supply and lymph circulation, as well as disorders of cell autoregulation, which leads to disruption of enzymatic processes in the cell.
Congenital disorders of enzyme systems (enzymepathies) are the cause of a large group of hereditary diseases - thesaurismoses (storage diseases), in which, due to the absence of any enzyme or its incorrect structure, any part of the metabolism is blocked, and abnormal substances appear in the cells.
The following morphogenetic mechanisms leading to the development of dystrophy are distinguished:: infiltration, decomposition (phanerosis), perverted synthesis, transformation.
Infiltration – this is the excessive penetration of metabolic products from the blood and lymph into cells or intercellular substance with their subsequent accumulation. A typical example of infiltration is the infiltration of the inner lining of the aortic wall (intima) and large vessels with cholesterol and its esters, which underlies a disease such as atherosclerosis.
Decomposition or phanerosis - this is the breakdown of cell ultrastructures and intercellular substance, leading to disruption of tissue or cellular metabolism and the accumulation of products of impaired metabolism in a tissue or cell, for example, with diphtheria intoxication, fatty degeneration of cardiomyocytes occurs. Perverted Synthesis is the synthesis of substances in cells and tissues that are not found normally. An example is amyloidosis, in particular in myeloma, when B lymphocytes, which normally produce antibodies, produce proteins with an altered structure during tumor transformation.
Transformation – the formation of products of one type of exchange instead of products of another type of exchange. For example, the transformation of fat and carbohydrate components into proteins.
Classification of dystrophies.
Depending on the structures in which dystrophic changes are predominantly localized, they are divided into parenchymal, stromal-vascular (mesenchymal), and mixed.
Depending on the predominance of disorders of one or another type of metabolism, protein, fat, carbohydrate, mineral, and mixed dystrophies are distinguished.
Acquired and hereditary dystrophies are also distinguished, and depending on the prevalence - general and local.
Parenchymal dystrophies
In these dystrophies, substances accumulate in the parenchyma cells of various organs, such as myocardiocytes, hepatocytes, neurons, cells of the renal tubules, and adrenal glands. Based on the type of metabolic disorder, these dystrophies are divided into protein (dysproteinoses), fat (lipidoses) and carbohydrate.
Parenchymal protein dystrophies (dysproteinoses) characterized by a violation of the exchange of cytoplasmic proteins in a free or bound state . These include granular, hydropic, hyaline-droplet and horny dystrophy.
Granular dystrophy .
The causes of granular dystrophy can be circulatory disorders, infections, intoxications and other factors that lead to a decrease in the intensity of the redox processes of the cell. In this process, the organs are enlarged, flabby, dull and cloudy when cut. This gives grounds to call granular dystrophy cloudy (dull) swelling of organs.
Microscopically, the cells are enlarged, their cytoplasm is cloudy and rich in protein granules. Electron microscopy with granular dystrophy shows an increase in the number (hyperplasia) and swelling of cell organelles, which optically look like protein granules. Such hyperplasia of cell ultrastructures is currently considered as a pronounced functional strain of organs to various influences.
The outcome of granular dystrophy is different. In most cases, it is reversible, but if the causes that caused it are not eliminated, it can transform into hyaline-droplet, hydropic or fatty degeneration.
The function of the affected organs may be weakened.
Hyaline droplet dystrophy .
It is a more severe type of dystrophy. Most often it develops in the kidneys, liver, and less often in the myocardium.
With this type of dystrophy, large drops of protein appear in the cytoplasm of cells, often merging with each other and, as a result of protein coagulation, resembling the ground substance of hyaline cartilage.
The appearance of organs is usually not changed or depends on the diseases in which this dysproteinosis occurs.
In the kidneys, such dystrophy develops in cases where a large amount of protein penetrates into the urine through the kidney filter. In this case, the accumulation of protein inclusions in the cytoplasm and its destruction are caused by the failure of lysosomal reabsorption of protein from the epithelium of the renal tubules under conditions of increased permeability of the glomerular filter (in nephrotic syndrome).
In the liver, hyaline droplet dystrophy often develops through perverted synthesis. Most often it develops with alcoholic damage, when drops of hyaline-like protein appear in the cytoplasm of hepatocytes, which are called Mallory bodies (or alcoholic hyaline) and are a morphological sign of alcoholic hepatitis. Sometimes hyaline-droplet degeneration of hepatocytes can be observed in biliary cirrhosis and Wilson-Konovalov disease. Alcoholic hyaline can have a cytolytic effect on hepatocytes and stimulate collagen synthesis, which determines the chronic progressive course of alcoholic hepatitis and contributes to the development of liver cirrhosis.
Organ function in hyaline droplet dystrophy is usually significantly affected. The outcome of this dystrophy can be coagulative necrosis (death) of the cell.
Hydropic (dropsy) dystrophy
This dystrophy is characterized by the appearance in cells of vacuoles filled with cytoplasmic fluid. It occurs due to disturbances in water-electrolyte and protein metabolism, which occurs during intoxication of various origins, as well as for infections, especially viral ones. Thus, it develops in the cells of the epidermis and mucous membranes during herpetic infection, chickenpox and natural pox; in hepatocytes – with viral hepatitis; in neurons of the brain and their processes - in spongioform encephalopathies (including Creutzfeldt-Jakob disease) caused by an infectious protein molecule - a prion. Vacuolar degeneration can affect the axial cylinders of the nerve fibers of the spinal cord conduction system in amyotrophic leukospongiosis and the myelin sheaths of nerve fibers in HIV infection (vacuolar myelopathy).
The appearance of the organs is usually not changed, with the exception of the skin during herpetic infection and smallpox, when bubbles (vesicles) filled with serous fluid appear.
On microscopic examination, the altered cells are increased in volume, their cytoplasm is filled with vacuoles of various sizes, and the nucleus is displaced to the periphery of the cell. As the process progresses, the vacuoles merge with each other and the cell turns into a vesicle, which consists of one large vacuole and a displaced vesicle-like nucleus ( balloon dystrophy)
In the kidneys, hydropic dystrophy of the convoluted tubule epithelium may be due to insufficiency of the reabsorption system of the basal labyrinth, which is overfilled with water penetrating the cell, and, “rising” to the brush border, destroys the membranes and forms balloon structures.
Hydropic degeneration of hepatocytes is most often the result of the action of the hepatitis B virus or toxic substances. Under the influence of a virus, this dysproteinosis occurs through perverted synthesis, subordinate to the reproduction of the virus, and under the influence of toxic substances, it is caused by insufficiency of the detoxification system.
Horny dystrophy
This condition is characterized by excessive formation of horny substance in stratified squamous keratinizing epithelium (skin), or keratinization of stratified squamous non-keratinizing epithelium (esophagus, cervix).
Horny dystrophy can occur with disorders of skin development, chronic circulatory disorders, chronic inflammation, infections, vitamin deficiencies, tumors, etc.
Horny skin dystrophy can be common ( ichthyosis- a hereditary disease manifested by excessive keratinization - excessive scaly peeling), and local ( hyperkeratosis).
Keratinization of stratified squamous non-keratinizing epithelium, for example, in the oral cavity or in the cervix, macroscopically looks like white spot (leukoplakia) .
Hereditary parenchymal dysproteinoses, which are caused by a violation of the intracellular metabolism of amino acids, include: cystinosis, tyrosinosis and phenylpyruvic oligophrenia (phenylketonuria). These diseases most often affect the liver, kidneys, bone marrow, spleen and nervous system.
Parenchymal fatty degenerations (lipidoses) ).
These dystrophies are characterized by impaired metabolism of fats, mainly neutral ones, in the cytoplasm of cells. The predominance of one or another mechanism for the occurrence of lipidosis depends both on the cause that caused it and on the structural and functional characteristics of the organ. Most often, the development of fatty degeneration is associated with hypoxia, which occurs with chronic diseases of the cardiovascular system, respiratory system, diseases of the blood system, as well as with the action of toxic substances (various infections and intoxications) and an increase in the level of fatty acids in the blood plasma (general obesity ).
Typically, fatty degeneration develops in the heart, liver, and kidneys.
Fatty degeneration of the myocardium can most often occur as a result of hypoxia or intoxication (diphtheria, alcohol, phosphorus poisoning, arsenic, etc.). In both cases, mitochondrial damage and a decrease in the intensity of fatty acid oxidation develop.
Tiny fat droplets appear in muscle cells (pulverized obesity), which, with increasing changes (small-droplet obesity), replace the cytoplasm. The process is focal in nature and is observed in groups of muscle cells along the venous knee of capillaries as well as small veins, which explains the peculiar appearance heart: yellow transverse stripes (“tiger” heart) are visible from the endocardium. The myocardium is flabby, the size of the heart can be increased, and the cavities expanded.
The outcome of parenchymal fatty degeneration of the myocardium depends on the degree of its severity. Initial changes are reversible, deep ones lead to severe dysfunction.
Fatty parenchymal degeneration of the liver is the most common.
It can occur when the level of fatty acids in the blood plasma increases (diabetes mellitus, general obesity), as a result of decomposition (with infections and intoxications), transformation (with chronic alcoholism), with malnutrition due to a lack of protein in food, with genetic defects of enzymes.
Small droplets of fat appear in the cytoplasm of hepatocytes (pulverized obesity), which merge into large droplets (small-droplet obesity). As a result, the droplet fills almost the entire cytoplasm, pushing the nucleus to the periphery of the cell (large droplet obesity). Macroscopically, the liver enlarges, becomes flabby, and acquires an ocher-yellow or yellow-brown color (“goose liver”).
Liver function decreases sharply. When the action of the damaging factor (for example, alcohol, more than 1 month) ceases, the normal structure of the cell can be restored. Large-scale obesity leads to the death of hepatocytes, a mesenchymal reaction and the development of fibrosis resulting in cirrhosis.
Fatty kidney degeneration most often develops in the epithelial cells of the proximal and distal tubules. by infiltration (with hyperlipidemia), less often - by decomposition (with increasing hypoxia). Most often, fatty kidneys occur in nephrotic syndrome. The kidneys are enlarged, flabby, with gray-yellow specks on the section. Kidney function decreases.
There is a group of hereditary systemic lipidoses, which are storage diseases (thesaurismoses). These diseases are associated with hereditary defects in enzymes that metabolize complex fats. These include: cerebroside lipidosis (Gaucher disease), sphingomyelin lipidosis (Niemann-Pick disease), ganglioside lipidosis (Tay-Sachs disease or early amaurotic idiocy), generalized gangliosidosis (Norman-Landing disease), etc. The liver, spleen, bone marrow and central nervous system. Morphological diagnosis is helped by cells found in tissues characteristic of a particular type of lipidosis (Gaucher cells, Pick cells)
Parenchymal carbohydrate dystrophies. Characterized by impaired glycogen or glycoprotein metabolism.
Glycogen metabolism disorders develop in diabetes mellitus. With this disease, there is an absolute and relative deficiency of insulin, as a result of which glucose utilization and glycogen synthesis are disrupted. As a result, the blood glucose level increases (hyperglycemia), glucose appears in the urine (glucosuria), and glycogen reserves in the liver and muscles are depleted. In connection with hyperglycemia and glycosuria, glucose infiltrates the tubular epithelium of the kidneys and glycogen synthesis occurs in the tubular epithelium, where it does not normally exist. The cytoplasm of kidney tubule cells becomes light and foamy.
Excessive accumulation of glycogen in the cells of the liver, kidneys, skeletal muscles also occurs in hereditary storage diseases (thesaurismosis) - glycogenosis caused by a deficiency of various enzymes that regulate glycogen metabolism (Gierke's, Andersen's, Pompe's, McArdle's diseases, etc.)
When glycoprotein metabolism is disrupted, mucins and mucoids (mucus-like substances) accumulate in cells. This is usually observed when inflammatory processes in mucous membranes, for example, with rhinitis, gastritis, bronchitis, etc. Such mucus can close the lumens of the bronchi and gland ducts, which leads to the formation of cysts. Sometimes mucus-like colloidal substances are produced in the thyroid gland during colloid goiter. The process ends with atrophy of the mucous membranes. Excessive accumulation of mucus is also observed in cystic fibrosis (a systemic hereditary disease). In this case, epithelial cells of the pancreatic glands, bronchial tree, digestive and urinary tracts, sweat and lacrimal glands, bile ducts) produce thick viscous mucus. The lumens of the glands become cystically enlarged, and connective tissue grows around them - cystic fibrosis.
STROMAL-VASCULAR (mesenchymal) DYSTROPHIES
Mesenchymal dystrophies are detected in the stroma of organs and walls blood vessels, and depending on the type of metabolic disorder, they are divided into protein, fat and carbohydrate.
TO mesenchymal dysproteinosis include: mucoid swelling, fibrinoid swelling, hyalinosis and amyloidosis.
At mucoid swelling in the main substance of connective tissue (usually in the walls of blood vessels, endocardium, synovial membranes) accumulation and redistribution of glycosaminoglycans occur, which have the property of attracting water, as well as plasma proteins, mainly globulins.
At the same time, collagen fibers swell and become unfibered, but remain preserved. Accumulating glycosaminoglycans have the phenomenon of metachromasia (the ability to change the basic color tone), which easily makes it possible to identify foci of mucoid swelling in the connective tissue. This phenomenon is most clearly manifested when stained with toluidine blue, when foci of mucoid swelling are colored not blue, but lilac or red.
This dystrophy most often develops in infectious-allergic (glomerulonephritis), allergic (immediate hypersensitivity reactions) and autoimmune (rheumatic diseases) diseases. The appearance of the organ is not changed, and mucoid swelling is detected only microscopically. When the cause is eliminated, it can be reversible, otherwise the disorganization of the connective tissue increases and fibrinoid swelling subsequently develops.
At fibrinoid swelling tissues continue to be impregnated with plasma proteins, which accumulate in the ground substance and collagen fibers, destroying them and turning them into a homogeneous mass containing fibrinoid, a complex substance consisting of fibrin, polysaccharides, immune complexes (for rheumatism), nucleoproteins (for systemic lupus erythematosus). Metachromasia of connective tissue is no longer detected, since the destruction of glycosaminoglycans of the main substance has occurred. Fibrinoid swelling is an irreversible process that can result in fibrinoid necrosis, sclerosis or hyalinosis.
At hyalinosis in the connective tissue, homogeneous translucent dense protein masses are formed, reminiscent of hyaline cartilage (hyaline).
Hyalinosis can be general and local. Hyalinosis of blood vessels and the connective tissue itself are also distinguished.
Hyalinosis can result from three conditions: fibrinoid swelling and necrosis, sclerosis and plasma impregnation.
Vascular hyalinosis affects small arteries and arterioles and occurs as a result of plasma impregnation in arterial hypertension and diabetes mellitus. Due to the deposition of blood plasma proteins in the wall, the vessels lose their elasticity, their lumen narrows, as a result of which the blood supply to the organ deteriorates. Vascular hyalinosis is a systemic process and is most pronounced in the kidneys, brain, retina, pancreas, and skin. Hyalinosis of cerebral arterioles is especially dangerous, since sudden rises in blood pressure (hypertensive crises) can lead to rupture of the vessel and hemorrhage into the substance of the brain.
Hyalinosis of the connective tissue itself develops as a result of fibrinoid swelling or sclerosis. As a result of fibrinoid swelling, hyalinosis occurs in diseases associated with immune disorders, for example, in rheumatic diseases and usually completes the progressive destruction of connective tissue. In rheumatism, hyalinosis occurs in the heart valves and chords, which contributes to the formation of heart defects.
As a result sclerosis hyalinosis forms in the pleura, pericardial layers, and peritoneum after suffering inflammatory processes. Hyalinosis of the spleen capsule leads to the formation of a “glazed spleen” - the capsule becomes thick, saturated with protein masses. Hyaline may also be deposited in connective tissue the skin after damage, especially after burns in the face and neck, which leads to the formation of rough, dense, whitish scars (keloid scars).
Amyloidosis.
Amyloidosis (Latin amylum - starch) is a stromal vascular dysproteinosis, accompanied by profound changes in protein metabolism and the appearance of abnormal fibrillar protein - amyloid.
In 1884, Rokitansky called amyloidosis a sebaceous disease, because. the affected organs have a greasy appearance. Later, R. Virchow showed that under the influence of iodine and sulfuric acid this substance turns blue and proposed calling it amyloid. The protein nature of amyloid was established in 1865 (M. Rudnev, Kuehne).
Microscopically, when stained with hematoxylin and eosin, amyloid appears as eosinophilic dense structureless masses. In order to distinguish amyloid from other deposits, histochemical methods are used, for example, Congo red, iodine, gentian violet and methyl violet staining, based on the phenomenon of metachromasia, since amyloid masses contain glycosaminoglycans.
The following main types of amyloid are distinguished depending on its chemical structure:
AA amyloid, which is detected in some types of hereditary (familial Mediterranean fever) and secondary amyloidosis. As a result of activation of the monocytic phagocyte system in the liver under the influence of interleukin-1, the synthesis of SAA is stimulated, which is subsequently degraded with the formation of protein AA from which amyloid fibrils are assembled on the surface of macrophages (amyloidoblasts);
AL amyloid, detected in primary amyloidosis and neoplastic plasma cell dyscrasia. First, the synthesis of light chains of immunoglobulins occurs from which amyloid fibrils are synthesized by plasma cells, macrophages and myeloma cells;
FAP (AF) amyloid is detected in some types of hereditary amyloidosis (familial amyloid polyneuropathy)
AS amyloid arising from senile amyloidosis
Amyloid formation is closely related to connective tissue fibers. Amyloid can be deposited along the reticular fibers (perireticular amyloidosis), for example, in the spleen, liver, kidneys, intestines, and along the collagen fibers (pericollagenous amyloidosis), for example, in striated and smooth muscles, skin.
Amyloidosis is also divided according to the predominant deposition of amyloid in organs into nephropathic, cardiopathic, neuropathic, hepapathic, etc.
The following clinical and anatomical forms of amyloidosis are distinguished:
Idiopathic (primary) amyloidosis. The cause and mechanism are unknown. In this case, amyloid appears in the myocardial stroma and vascular walls (cardiopathic amyloidosis), as well as along the peripheral nerves (neuropathic amyloidosis)
Hereditary (genetic, family) amyloidosis. These are cardiopathic, neuropathic, less often - nephropathic options. Most common in Mediterranean countries (Israel, Lebanon, etc.)
Senile amyloidosis. Amyloid is deposited in the cerebral cortex, being the center of senile (senile) plaques, as well as in the wall of small blood vessels. Such changes appear in large numbers in senile (senile) dementia and Alzheimer's disease.
Acquired (secondary) amyloidosis. It occurs most often and is considered as a complication of various diseases accompanied by chronic suppurative and destructive processes. Amyloid is deposited against the background of any disease: tuberculosis; chronic purulent inflammation, For example, nonspecific diseases lungs (bronchiectasis, chronic pneumonia), osteomyelitis, chronic abscesses, chronic sepsis, as well as rheumatic diseases (primarily rheumatoid arthritis); and blood tumors ( multiple myeloma). Amyloid is usually deposited in the kidneys, spleen, liver, adrenal glands, and intestines.
The organs increase in size, become dense, and have a greasy appearance when cut.
In the spleen, amyloid is deposited first in the lymphatic follicles in the form of translucent grains - sago spleen, then accumulates in the red pulp, the spleen enlarges, becomes dense, the cut surface is smooth, shiny - sebaceous spleen.
Kidney amyloidosis is of great importance in the clinic, as it is life-threatening for the patient. Beginning in the renal medulla pyramids, amyloidosis gradually invades the cortex. Amyloid is deposited in small vessels, in the mesangium of the glomeruli, on the basement membrane of the tubules, and in the stroma of the organ.
The bud increases in size, its substance is dense, white, with a greasy sheen (“large greasy bud”). A clinical and morphological picture of amyloid nephrosis emerges, which leads to chronic renal failure.
The liver with amyloidosis is also enlarged, dense, and looks “greasy.”
Less commonly, amyloid is deposited in the adrenal glands, usually in its cortex, and in the intestine, in the submucosal layer.
The functional significance is determined by the degree of development of amyloidosis. Severe amyloidosis leads to degeneration and atrophy of the parenchyma and sclerosis of the organ stroma, leading to their functional failure. With severe amyloidosis, chronic renal failure is most often observed, less often - liver, cardiac, pulmonary, adrenal, intestinal (malabsorption syndrome) failure.

What would New Year be without champagne, tangerines, Olivier, aspic and everyone’s favorite “Herring under a fur coat”. With the last one...

Let's prepare the necessary ingredients for the cookies. The first thing to do is put the water to boil. We need...

Is it possible to register an employee for the position of financial director - chief accountant? The chief accountant claims that yes, but...

The head of a small business can easily manage the budget independently. CHECKED! If you're budgeting...

The creation of new projects involves a preliminary economic justification for their feasibility, subsequent...
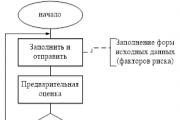
Reporting is generated by the RM, is agreed upon (approved) by the Risk Committee under the Management Board and transmitted to...

At the edge of a large, very large meadow, on a long emerald blade of grass lived a tiny Ladybug. God's little...

Nowadays, it is quite common for people to turn to the stars. With the help of a horoscope a person can find out...

Business or friendly. If you were pursuing a stranger, it means your level of trust in the female sex...

according to Freud's dream book If you dreamed about how you were fishing, it means that in real life you can hardly switch off...

The New Year's whirlwind will spin us around so quickly and rapidly that it's time to thoroughly think through the New Year's...

The article offers you some of the most delicious recipes for making vinaigrette. Vinaigrette is a delicious salad...

July 4th, 2015 , 08:33 pm Lately our Nastena has been completely giving up sweets (and thank God), but...

Fragrant, very tasty chocolate cake, it’s impossible to stop eating! The Negro’s Kiss cake is very easy to prepare,...

Let's prepare the necessary ingredients for the cookies. The first thing to do is put the water to boil. Us...

Is it possible to register an employee for the position of financial director - chief accountant? The chief accountant claims...