Philosophical vision of the problem
The meaning of life is related to the question “What to live for”, and not to the question of how to maintain life. A person's attitude is...
One of the rare and extremely dangerous diseases for human health and life is amyotrophic lateral sclerosis. This is a pathology of the nervous system in which motor neurons spinal cord, as well as the cortex and brain stem, undergo irreversible changes. The disease is chronic and characterized by constant progression. The pathological process is not very common: approximately 4-6 cases of the disease are recorded per 100,000 people.
Amyotrophic lateral sclerosis (ALS) is a disorder in the functioning of the nervous system that provokes constantly progressive muscle weakness, which occurs under conditions of selective damage to motor neurons. The pathology has several other names, for example, Lou Gehrig's disease, named after the world champion baseball player who suffered from this disorder. Another name for side amyotrophic sclerosis- Charcot's disease. It is associated with the name of the French psychiatrist Jean-Martin Charcot, who described pathological muscle weakness in the second half of the 19th century.
Finally, ALS is known as motor neuron disease according to ICD-10.

Sometimes, when describing this pathological condition, the word “lateral” is replaced by the epithet “lateral”. The inclusion of this adjective in the name of the disease is due to the fact that the neurons located in the lateral projections of the spinal cord are most susceptible to changes.
Pathological changes associated with muscle weakening and atrophy are caused by the destruction of neurons that are responsible for transmitting signals from the brain to the muscles. With this disease, both central and peripheral motor neurons can be affected. The first of them is located in the cortex cerebral hemispheres brain When it is damaged, muscle weakness develops, while tone increases and reflexes become stronger. The peripheral motor neuron is located in the brainstem and on various levels spinal cord. If it undergoes pathological changes, then the development of muscle weakness is observed, but at the same time reflexes decrease, as does muscle tone, as well as the development of muscle atrophy.
In conditions of damage to one of the motor neurons (or both at once), the transmission of impulses from it to the muscle is blocked.
Amyotrophic lateral sclerosis manifests itself in increasing muscle weakness and muscle wasting, which, as a result, chronic course, leads to complete immobility of the patient and impaired respiratory functions.

Currently, there are about 70,000 people in the world diagnosed with ALS. Usually this pathology manifests itself after the age of 40-50 years. According to clinical data, it usually leads to death within 5 years after diagnosis. However, the famous physicist Stephen Hawking has been living with this disease for 50 years.
Only 7% of all patients live more than 60 months.
It has been established that men suffer from Charcot's disease more often than women. Recently, scientists have suggested that greatest number cases of pathology are recorded in people with high intelligence and athletes who have not had serious health problems throughout their lives.
Amyotrophic lateral sclerosis became known to medicine not too long ago, and the main cause of this disease is still not clear. However, researchers agree that it is based on the accumulation of pathological insoluble protein in the motor cells of the nervous system. This is what leads to their death.
There is a theory according to which the leading role of development irreversible changes belongs to a change in the properties of a special enzyme, the function of which is to protect the body’s cells from destruction by oxygen radicals. This enzyme is called superoxide dismutase-1. Such changes are associated with gene mutations, which in 25% of cases are inherited. It is this fact that motivates the hypothesis about genetic nature BASS.
It is possible that the destruction of motor neurons is associated with mutations in other structures, for example, formations that give the nerve cell its shape and provide its framework.
When describing the nature of amyotrophic lateral sclerosis, they mention following reasons its development:
Hosts of the program “Live Healthy!” will talk about incurable disease more details:
Doctors include the following risk factors:
Amyotrophic lateral sclerosis is not infectious disease, they cannot be infected from another person.
Motor neuron disease is classified into types based on the severity of damage to nerve cells. The manifestations of each of them are basically similar, but as they develop pathological process, the difference becomes more obvious.
In neurology there are following forms diseases:

The photo shows progressive bulbar palsy
The progression of amyotrophic lateral sclerosis causes complications that lead to the death of the patient. These should include:
The disease begins with damage to the limbs, which then spreads to the rest of the body.
Early symptoms of amyotrophic lateral sclerosis are:
Dear readers, about the main causes and symptoms of the disease, watch the video below:
As amyotrophic lateral sclerosis progresses, the patient notices muscle stiffness, which is associated with an increase in their tone and difficulty in trying to relax them. There are also such characteristic features, such as imbalance, spontaneous bouts of laughter or crying, limited tongue movements, change in voice.
In later stages of the disease, interruptions in breathing occur, depression, and the inability to move independently are noted.
Diagnosis of amyotrophic lateral sclerosis requires a large number of specific measures. First of all, the patient is examined and interviewed by a neurologist who is interested in the following information:
Other methods that allow you to make a correct diagnosis include:

The procedure for performing a lumbar puncture involves piercing the arachnoid membrane of the spinal cord between the 3rd and 4th, or 2nd and 3rd lumbar vertebrae with a Beer needle to collect cerebrospinal fluid.
In addition to the listed methods, differential diagnosis must be made. Amyotrophic lateral sclerosis must be distinguished from pathologies such as spinal cord tumor, cervical myelopathy, malabsorption syndrome, and diabetic amyotrophy.
Treatment of amyotrophic lateral sclerosis aims to slow down the progression of the pathological process, since the changes that occur are irreversible. The disease cannot be cured.
The main and only drug currently recommended to slow the progression of the disease is Riluzole, or Rilutek. Its main active ingredient prevents further damage to neurons, thus slowing down the progression of the disease.
Other drugs are used solely to relieve prevailing symptoms that impair the patient's quality of life. These should include:

Different types of action of muscle relaxants
A patient diagnosed with ALS requires devices that allow him to move (wheelchair, walker, bed with various functions, in particular, equipped with a lift). In order for the patient to be able to breathe, a special operation can be performed - tracheostomy. During manipulation in the trachea surgically create a hole.
The patient must be provided complete care. Particular attention should be paid to measures to prevent the formation of bedsores. The bed should always be dry and clean, as should the patient’s body.
Another integral component of maintenance therapy for amyotrophic lateral sclerosis is working with a psychologist. The help of a specialist will be needed not only by the patient, but also by his family members.

Due to disability due to ALS, the patient is required to use a wheelchair
The prognosis for this specific disease is poor. The disease constantly progresses, increasingly worsening the patient's quality of life and ultimately leading to acute respiratory failure and death of the patient. Depending on the form in which the pathological process occurs, the patient can live from 2 to 12 years. With the bulbar form, as well as if the patient is elderly, life expectancy is reduced to 1-3 years.
Amyotrophic lateral sclerosis is a rare disease that constantly progresses and inevitably leads to the death of the patient. The reasons for this phenomenon have not been fully studied; there are only assumptions regarding probable risk factors. Treatment of ALS comes down to alleviating the patient’s condition and providing him with the most comfortable living conditions.
Amyotrophic lateral sclerosis (ALS), also called motor neuron disease or Charcot-Kozhevnikov, motor neuron disease, and in some places around the world Lou Gehrig's disease, which mainly affects language-speaking regions. English language. Dear patients, in this regard, should not be surprised or doubt if in the text of our article they come across various names for this very bad pathological process, leading first to complete disability and then to death.
The basis of this terrible disease are lesions of the brain stem, which do not stop in this area, but spread to the anterior horns of the spinal cord (the level of the cervical thickening) and pyramidal tracts, leading to degeneration of skeletal muscles. In histological preparations, cytoplasmic inclusions called Bunin bodies are found, and against the background of vascular infiltrates, degenerative changes, wrinkled and dead are observed nerve cells, in place of which glial elements grow. It is obvious that the process, in addition to all parts of the brain and spinal cord (cerebellum, brainstem, cortex, subcortex, etc.), motor nuclei of the cranial nerves (cranial nerves), affects meninges, cerebral vessels and spinal vascular bed. During the autopsy, the pathologist notes that the cervical and lumbar thickening in patients is noticeably reduced in volume, and the trunk is completely atrophied.

If 20 years ago patients could barely live 4 years, in our time there has been an increasing trend average duration life, which already reaches 5-7 years. The cerebral form still does not have longevity (3-4 years), and the bulbar form does not offer much of a chance (5-6 years). True, some live for 12 years, but mainly these are patients with the cervicothoracic form. However, what does this period mean if Charcot’s disease (sporadic forms) does not spare children (high school) and adolescence, while the male sex has a greater “chance” of acquiring motor neuron disease. Familial cases appear more often in adulthood. The real danger of getting sick remains between the ages of 40 and 60, but after 55 men no longer hold the lead and get sick just like women.
Bulbar disturbances in the activity of the centers responsible for respiratory function and work usually lead to death. of cardio-vascular system.
In the literature you can find such a definition as “ALS syndrome”. This syndrome has nothing to do with motor neuron disease, is caused by completely different reasons and accompanies other diseases (some proteinemia, etc.), although the symptoms of ALS syndrome are very reminiscent of the early stage of Lou Gehrig’s disease, when the clinic has not yet developed rapidly. For the same reason initial stage Amyotrophic lateral sclerosis is differentiated from () or.
 ALS has no boundaries in the sick human body, it moves further and, thus, affects the entire body of the patient, therefore the forms of amyotrophic lateral sclerosis are distinguished rather conditionally, based on the beginning of the pathological process and more clear signs defeats. Exactly predominant symptoms during amyotrophic lateral sclerosis, and not isolated affected areas, allow us to determine its forms, which can be presented in the following form:
ALS has no boundaries in the sick human body, it moves further and, thus, affects the entire body of the patient, therefore the forms of amyotrophic lateral sclerosis are distinguished rather conditionally, based on the beginning of the pathological process and more clear signs defeats. Exactly predominant symptoms during amyotrophic lateral sclerosis, and not isolated affected areas, allow us to determine its forms, which can be presented in the following form:
The factors that can trigger this severe pathological process are not so numerous, but a person can encounter any of them every day, regardless of age, gender and geographical location, except, of course, hereditary predisposition, which is typical only for a certain part of the population (5-10%).
So, the causes of motor neuron disease:
Symptoms of amyotrophic lateral sclerosis are characterized primarily by the appearance of peripheral and central paresis hands, as indicated by the following signs:

It is obvious that involving the entire body in the process, Charcot's disease gives rich and diverse symptoms, which, however, can be briefly represented by syndromes:
As for diagnostics, it relies primarily on neurological status, and the main instrumental method ENMG (electroneuromyography) is recognized to search for ALS; other testing is carried out to exclude diseases with similar symptoms or to study the patient’s body, in particular, the state of the respiratory system and musculoskeletal system. Thus, the list necessary research includes:
Therapy for motor neuron disease is primarily aimed at general strengthening, maintaining the body and relieving symptoms. As the pathological process develops, respiratory failure increases, so in order to improve respiratory activity, the patient first (while still in a wheelchair) switches to a NIV device (for non-invasive ventilation of the lungs), and then, when he can no longer cope, to stationary ventilator equipment.

For real effective remedy has not yet been invented for the treatment of amyotrophic lateral sclerosis, however, treatment is still necessary and the patient is prescribed drug therapy:
One can hardly argue with the statement that a patient with Charcot's disease needs special care. It’s special, because feeding alone is worth it. What about the fight against bedsores? What about depression? The patient is critical of his condition, is very worried that every day his condition is worsening and, ultimately, he stops (not of his own free will) to take care of himself, cannot communicate with others and enjoy a delicious dinner.
Such a patient needs:
Prevention of bedsores is very important. They're in similar cases do not keep yourself waiting long, so the bed should be clean and dry, as well as the patient’s body.
The patient eats mainly liquid, easily swallowed food, rich in proteins and vitamins (as long as swallowing function is preserved). Subsequently, the patient is fed through a tube, and then they resort to a forced, but last measure - the imposition of gastrostomies.

It is obvious that a patient with amyotrophic lateral sclerosis suffers greatly: both morally and physically. At the same time, the people caring for him, for whom he is a close person, also suffer. Agree, it is very difficult to look into fading eyes, see pain and despair and not be able to help defeat the disease, cure it, bring it back to life. dear person. Relatives caring for such a sick person lose strength and often become despondent and depressive state, and therefore they also need the help of a psychotherapist with the prescription of sedatives and antidepressants.
Usually, in a description of the treatment of any disease, readers look for preventative measures and ways to get rid of a particular ailment folk remedies. Actually recommended alternative medicine, containing large amounts of B vitamins, wheat germ and oat grains, walnuts and propolis may not harm the patient, but they will not cure him either. In addition, we should not forget that such people often have problems with swallowing, so in the case of Charcot's disease, you should not rely on traditional medicine.
This is what it is - amyotrophic lateral sclerosis (and many other names for it). The disease is terribly insidious, incomprehensible and incurable. Maybe someday a person will be able to tame this disease, at least, let's hope for the best, because scientists all over the world are working on this problem.
Amyotrophic lateral sclerosis (ALS) is an incurable, progressive disease of the central nervous system in which the patient experiences damage to the upper and lower motor neurons, which causes muscle atrophy and paralysis. The frequency of this pathology is about 2-7 cases per 100 thousand people. Most often, the disease is diagnosed in patients over 50 years of age.
Scientists have not yet created a unified comprehensive classification of ALS. There are several approaches to classifying the disease. For example, the North American approach involves identifying the following types of ALS: sporadic, familial, sporadic endemic. The classification of amyotrophic lateral sclerosis provides for the following forms of the disease: bulbar, lumbosacral, cervicothoracic and primary generalized. There are also several variants of the disease: mixed, pyramidal and segmental-nuclear.
To the most common initial symptoms diseases include cramps (painful muscle spasms), lethargy and weakness in the distal arms, bulbar disorders, leg muscle atrophy, weakness in shoulder girdle. In addition, for different options The disease is characterized by various clinical manifestations.
The exact causes of amyotrophic lateral sclerosis are still being investigated by scientists. However, several factors can be named that provoke the disease. For example, about 5% of diseases have a hereditary etiology. At least 20% of cases are associated with mutations in the superoxide dismutase-1 gene. Scientists have proven that important role High activity of the glutamatergic system plays a role in the onset of the disease. The fact is that excess glutamic acid provokes overexcitation and sudden death of neurons. The molecular genetic mechanism of the pathology has also been proven. It is caused by an increase in the level of DNA and RNA in cells, which ultimately leads to disruption of protein synthesis.
Scientists also identify several predisposing factors that play an important role in the occurrence of ALS. First of all, such factors include age. The fact is that the disease usually develops in patients aged 30-50 years. It is worth remembering that only about 5% of patients have a hereditary predisposition to ALS. In the vast majority of cases of ALS, the cause of the pathology cannot be determined.
The early course of the disease is characterized by symptoms such as convulsions, twitching, muscle numbness, difficulty speaking, and weakness in the limbs. Since such symptoms are characteristic of many neurological diseases, diagnosing ALS at an early stage is difficult. In most cases, the disease can be diagnosed at the stage of muscle atrophy.
Depending on the disease affected different parts body, distinguish limb ALS and bulbar ALS. In the first case, patients experience deterioration in ankle flexibility, awkwardness when walking, and they begin to stumble. Bulbar ALS is manifested by difficulty speaking (nasal sound, difficulty swallowing). Soon the patient finds it difficult to move or can no longer move independently. Usually the disease does not have a detrimental effect on mental capacity patient, but leads to severe depression. In most cases, about three to five years pass from the appearance of the first symptoms to death.
Since ALS is an incurable disease that rapidly shortens a person’s life, the patient’s examination must be comprehensive and accurate. It is extremely important to put correct diagnosis the patient in order to begin timely relief of his main symptoms, as this can prolong the patient’s life. The examination plan usually includes a history of life and illness, a neurological and physical examination, MRI of the spinal cord and brain, EMG, and laboratory tests.
Diagnosis of the disease begins with a detailed interview with the patient. Namely, the doctor needs to clarify whether the patient complains of muscle spasms and twitching, weakness and stiffness, impaired movement of the hands, speech, walking, swallowing, salivation, frequent shortness of breath, weight loss, fatigue, shortness of breath during exercise. In addition, the doctor should ask whether the patient has noticed double vision, memory loss, crawling sensations on the body, or urinary problems. It is imperative to ask the patient about his family history - whether he has any relatives with chronic movement disorders.
The main purpose of the physical examination is to assess the patient's constitution, weigh him, measure his height, and calculate his body mass index. The neurological examination usually includes neuropsychological testing. When assessing bulbar functions, the doctor pays attention to the timbre of the voice, speed of speech, pharyngeal reflex, the presence of tongue atrophies, and paresis of the soft palate. In addition, during the examination, the strength of the trapezius muscles is checked.
The main instrumental method for diagnosing the disease is needle EMG. This technique allows you to identify signs of the disease such as acute or chronic denervation. In the early stages of the disease, stimulation EMG is ineffective because it does not detect noticeable signs of ALS.
In the process of diagnosing the disease, doctors also use neuroimaging methods. MRI of the spinal cord and brain plays a great role in the differential diagnosis of ALS. During MRI, in 17-67% of patients it is possible to identify symptoms of degeneration of the pyramidal tracts and atrophy of the motor cortex of the brain. However, it is worth noting that this technique is ineffective when diagnosing the disease in patients with bulbar syndrome.
Many laboratory tests are performed during the diagnosis of ALS. In particular, doctors can prescribe clinical and biochemical tests blood, cerebrospinal fluid examination, serological studies. However, the only effective and reliable method of analysis is still considered to be molecular genetic analysis. The presence of mutations in the superoxide dismutase-l gene is considered a suspicion for ALS.
Since the symptoms of amyotrophic lateral sclerosis are similar in many respects to the manifestations of other neurological pathologies, doctors must carry out differential diagnosis. The most accurate diagnosis can be made using MRI of the brain and spine. First of all, ALS must be differentiated from muscle diseases, which include Rossolimo-Steinert-Kurshman dystrophic myotonia, myositis with cellular abnormalities, and oculopharyngeal myodystrophy.
It is also necessary to distinguish ALS from spinal cord pathologies:
Differential diagnosis is also necessary in order to distinguish the disease from systemic pathologies, lesions of the neuromuscular synapse, and brain pathologies such as multiple system atrophy, dyscirculatory encephalopathy, and syringobulbia.
The main goals of treatment for amyotrophic lateral sclerosis are considered to be to slow the progression of the disease, as well as eliminate its symptoms, which significantly worsen the patient’s quality of life. It should be remembered that ALS is a serious incurable disease that shortens a person’s life expectancy. That is why the doctor has the right to inform the patient of the diagnosis only after a comprehensive and thorough examination.
Treatment of the disease includes drug and non-drug therapy. The latter implies security measures. The patient should limit physical activity, which can accelerate the progression of ALS. In addition, it is very important to eat properly and nutritiously. Drug therapy is divided into two types: pathogenetic and palliative.
To date, the only drug that can slow the progression of ALS is riluzole. It has been proven that taking it can prolong the patient’s life by an average of three months. This drug is indicated for patients whose disease duration is less than 5 years. The patient should receive 100 mg of the drug daily. To avoid the risk of drug-induced hepatitis, every three months it is necessary to check the levels of AST, ALT and LDH. Since men and smokers have lower concentrations of riluzole in their blood, they should either limit their smoking or quit smoking altogether. bad habit. You will need to take the drug for life.
Scientists have repeatedly tried to use other drugs for pathogenetic therapy. However, such experiments did not prove effective. Among them were:
The effectiveness of taking it has also not been proven high doses Cerebrolysin, despite the fact that this drug is able to slightly improve the condition of patients.
Palliative therapy is intended to eliminate a complex of symptoms of the disease and thereby improve the patient’s quality of life. To eliminate certain symptoms of ALS, the following techniques are used:
To improve muscle metabolism, a patient with ALS can be prescribed the following medications: creatine, carnitine, levocarnitine solution, trimethylhydrazinium propionate. Multivitamin therapy is also indicated for patients, which involves taking multivitamins (neuromultivit, milgamma) and thioctic acid.
In most patients with ALS, the disease is accompanied by serious motor impairments, including limited mobility. Of course, this causes great discomfort to the patient, who constantly needs help from other people. Eliminate some movement disorders Orthopedic correction techniques help. The doctor must explain to the patient that the use of assistive devices does not indicate his disability, but only reduces the difficulties caused by the disease.
The most life-threatening symptom of the disease is considered to be respiratory failure. Its earliest symptoms will be morning fatigue, vivid dreams, daytime sleepiness, and dissatisfaction with sleep. To detect respiratory failure at an early stage, polysomnography and spirography are performed. To eliminate apnea, medication and non-invasive ventilation are indicated. It has been proven that these techniques can prolong a patient’s life by one year. If the patient needs assisted breathing for more than 20 hours, the doctor raises the question of a complete transition to invasive ventilation.
Patients who have undergone an initial examination or a repeated diagnosis of the disease must remain under outpatient observation. As any new symptoms appear, they should also receive qualified advice. Patients must take most medications regularly. Only vitamins and myotropic drugs are taken in courses in stages.
Every three months the patient must undergo spirography. If he takes riluzole regularly, he needs to have LDH, AST, and ALT checked every six months. If the patient has dysphagia, blood glucose levels and trophic status should be measured periodically. Patients have a choice of treatment regimen: they can either stay at home or stay in a hospice.
The prognosis for patients with ALS largely depends on the course of the disease. It has been proven that about 80-90% of patients who experience severe respiratory complications die within 3-5 years after the first signs of the disease appear. The remaining 10% of patients have a benign course of the disease. The duration of the disease is significantly reduced in the presence of the following factors: the patient’s age is less than 45 years, bulbar onset of ALS, rapid progression of the disease.
Amyotrophic lateral sclerosis is a serious neurological disease that causes muscle weakness, disability and ultimately death. ALS is often called Lou Gehrig's disease, after the famous baseball player who was diagnosed in 1939. In some countries, ALS and motor neurone disease are sometimes used interchangeably.
Worldwide, ALS affects 1-3 people per 100,000. In the vast majority of cases - from 90 to 95 percent of cases of this disease, doctors cannot explain the cause of this disease. Only in 5-10 percent of cases is genetic determination traced. ALS often begins with muscle cramps in an arm or leg and difficulty speaking. Ultimately, ALS disrupts control of the muscles responsible for the breathing movements of swallowing.
Early symptoms of ALS include:
The disease often begins in the arms, legs, or limbs and then spreads to other parts of the body. With the onset of the disease, symptoms begin to progress, the muscles become weaker and then paralysis occurs. Ultimately, there is a violation of the acts of chewing, swallowing and breathing.
With amyotrophic lateral sclerosis, the nerve cells that control movement (motor neurons) begin to gradually die, which leads to a gradual weakening of the muscles and their atrophy. ALS is inherited in 5-10 percent of cases. In other cases, ALS appears to occur spontaneously.
Several possible causes of ALS are currently being studied, including:
Major risk factors for ALS include:
Environmental factors that increase the risk of this disease include:
 As the disease progresses, patients with amyotrophic lateral sclerosis experience the following complications.
As the disease progresses, patients with amyotrophic lateral sclerosis experience the following complications.
If some of the early symptoms of neuromuscular diseases appear, you should contact your doctor, who, if necessary, will refer you for a consultation with a neurologist. But even a timely visit to a neurologist does not guarantee that the diagnosis will be made immediately, since it takes some time to verify the diagnosis. The neurologist will be interested in the medical history and neurological status.
Amyotrophic lateral sclerosis is quite difficult to diagnose in the early stages, since the symptoms are similar to other neurological diseases. The following diagnostic methods are used:
Due to the fact that the processes in amyotrophic lateral sclerosis cannot be reversed, treatment is aimed at slowing the progression of symptoms.
Drug treatment. The drug riluzole (RILUTEK) is the first and only drug approved to slow ALS. The drug has an inhibitory effect on the progression of the disease in some patients, possibly by reducing the level of glutamate, a substance that is a mediator in the nervous system and the level of which is often elevated in patients with ALS. In addition, other medications may be prescribed to relieve symptoms such as constipation. muscle cramps fatigue hypersalivation pain depression.
Exercise therapy. Physical exercises under the supervision of a physical therapy doctor can help maintain muscle strength and range of motion for more a long period activity of the cardiovascular system and improve overall well-being.
Using a walker or wheelchair also allows you to maintain a certain range of movements.
Psychological help. The help of a psychologist is often required due to the patient’s awareness of the incurability of the disease. Although in some cases life expectancy can exceed 3-5 years and reach 10 years.
ALS syndrome stands for. The disease affects the central nervous system, develops slowly, but attacks the spinal cord and brain, as well as the nuclei of the cranial nerves. If left untreated, the disease can damage motor neurons, causing paralysis and muscle atrophy. Let's look at what myelopathy is and what its symptoms, causes, and how treatment is carried out.
Myelopathy is a spinal cord pathology that can develop in ALS syndrome. If the functioning of the spinal cord is disrupted, functions and reactions are insufficient, a person faces dire consequences.
Reasons that provoke the appearance of myelopathy:
The disease can develop due to one of these factors, or due to several.
Myelopathy has the following symptoms:

The listed symptoms require therapy. But before starting treatment, it is important to diagnose myelopathy. Modern equipment and differential tests are used to make a diagnosis.
Myelopathy requires treatment. Disease in acute period treated by eliminating pain in the affected area. Treatment is carried out using a blockade. The skin around the lesion is injected with painkillers. Thanks to the blockade, the brain does not receive a signal about the presence of inflammation in the joints or muscles. As a result, pain is blocked for a long time.
After this, treatment is carried out using:
Thanks to these measures, the patient’s well-being improves.
Very often myelopathy develops into chronic form. In this case, to prevent the disease from progressing and exacerbating, it is important to maintain the disease in a state of remission.
In order for the disease to bother you as little as possible, it is important to carry out its prevention.
Preventive measures are aimed at preventing the causes that cause a decrease in the functions of the spinal cord. 
You need to eat right, toughen up, try to get rid of excess weight. To prevent any disease, strengthen your immune system in every way.
For children, to prevent illness, create a schedule of work and rest, and distribute the load.
Due to the mutation of proteins with the development of intracellular aggregates, the disease ALS begins to form. The disease usually affects people between 40 and 60 years of age.
The exact causes of the development of the disease are not fully understood. But scientists have found that the disease develops due to the appearance of four-stranded DNA in human cells. As a result, protein synthesis is disrupted and symptoms of the syndrome begin to appear.
To make an accurate diagnosis, it is important to visit several specialists.
5 percent of people acquire the disease from their relatives due to a hereditary factor.
The causes of the syndrome can be such as infection, injury, or infectious diseases.
Most people today have symptoms of ALS syndrome. In each case, the disease has its own causes and symptoms of development. It is important to monitor your health in order to identify pathology in the early stages of manifestation and begin its treatment. 
Symptoms of the syndrome:
ALS disease is very difficult to diagnose in the early stages. But experienced specialists study the symptoms, consider different versions diseases, and only after that a diagnosis is made and treatment is prescribed.
If the patient is nevertheless diagnosed with ALS, his relatives need to prepare for difficulties.
The patient completely loses the ability to move independently. Then he cannot eat normally. Also sometimes there is very strong drooling. Some patients require special enteral nutrition.
After some time, problems with the respiratory system begin and respiratory failure develops.
Patients are often bothered by headaches, suffocation, and nightmares. 
Taking into account the location of the muscles, experts identify the following forms of ALS syndrome:
The bulbar form of the disease is characterized by the following disorders: numbness of the tongue, paralysis of the palate, weakness of the masticatory muscles. Speech and the ability to swallow normally are also impaired. IN severe cases the patient may begin to laugh or cry for no reason, move animatedly lower jaw. After some time, the arms and legs are affected. Most often, even after proper treatment, patients die.
In the cervicothoracic form of the disease, the muscles of the arms and legs atrophy.
With the lumbosacral form, atrophic paresis of the lower extremities occurs. When the form is advanced, the arms and cranial muscles are also paralyzed.
In the cerebral form, the limbs are paralyzed and peripheral motor neurons are affected. The patient laughs and cries for no reason. Sometimes he begins to actively move his lower jaw.
To make a diagnosis, an electromonogram is performed. The study shows that there is a “picket fence” rhythm in fasticulation potentials; the conduction speed does not change. To make a correct diagnosis it is important to follow all areas of the spine:

It is important not to confuse the syndrome with other types of disease that may have similar symptoms. Therefore, an accurate diagnosis must be made by experienced specialists.
Treatment for ALS aims to relieve symptoms of the disease. Therapy is carried out with Riluzone. But, this drug is available only in Europe and the USA. Medicine does not cure the disease. But, it helps to prolong and improve the quality of life of a sick person.
Treatment of ALS syndrome is carried out with the following drugs:
Riluzon's work. When is it transmitted? nerve impulse, glutamate, a chemical mediator in the central nervous system, is released. Rizulon reduces the amount of excretion of such a substance.
After research, scientists found that excess glutamate damages neurons in the spinal cord and brain. According to tests, people who use Rizulon live longer than other patients - by three months.
Scientists have also found that antioxidants suppress the signs of the syndrome. These substances help the body prevent free radical damage. Antioxidants are selected by the doctor taking into account the patient’s health condition. 
To make life easier for people with the syndrome, concomitant treatment. Since the disease takes a very long time to be treated, it is important to treat not only the main disease, but also other symptoms. According to experts, relaxation relieves anxiety and fear.
Reflexology, aromatherapy and massage are used to relax muscles. Thanks to such procedures, lymph and blood circulation is normalized and pain is eliminated. By performing all procedures, specialists stimulate endogenous painkillers and endorphins. But it is important to carry out activities with nervous system individually. Therefore, before starting treatment, visit a doctor and undergo all examinations.
The syndrome is constantly progressing and if left untreated, it can be fatal. Once signs of the disease are detected, the patient has the opportunity to live another five years. But in order for his life to proceed well, provide supportive therapy.
An unfavorable sign is age over 50 years, as well as the development of abnormalities in the functioning of the human body.
Now you know what the essence of ALS syndrome, as well as myelopathy, is. Why the disease occurs, what forms and symptoms exist, and also what methods of therapy are carried out. Since the syndrome is deadly, it is important to show the patient to a specialist at the first signs in order to carry out the necessary therapy in order to prolong and improve the life of the sick person.

The meaning of life is related to the question “What to live for”, and not to the question of how to maintain life. A person's attitude is...

A mushroom is a living organism that forms a separate kingdom of the same name. For a long time they were classified as part of the plant kingdom. But in...

For lovers of “quiet” hunting, mushroom season begins in early summer and lasts until late autumn. And rarely do they...

Aleshnikova, V.I. Use of professional consultants. - M.: Infra-M, 1999. - 240 p. 2. Beich, E....

Orange juice. The symbolic meaning of orange juice in dream books is pleasure and temptation. Quite often we...

We most often overcome all kinds of difficulties encountered along the path of life. Of course, for this we make...

This year your patron Neptune will be in your constellation and this is a good sign, because you will...

1993 who? 1993 is the year of which animal? — According to the Chinese horoscope, 1873, 1933, 1993 belonged to the years of the Black...

Wave-particle duality of light means that light simultaneously has the properties of continuous...

The role of biology is enormous in our world. Although it is not one of the priority subjects, most schoolchildren and...
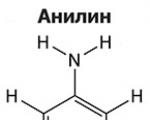
Amines are organic derivatives of ammonia containing an NH 2 amino group and an organic radical. In general...
How to answer questions in Part BThe second part of the social studies work consists of 7 tasks with a short answer....

Form TORG-15 is drawn up in the case when during transportation, movement between and within the warehouse, when...

Nutritionists say that for good health and a slim figure, you must include snacks in your...

A mushroom is a living organism that forms a separate kingdom of the same name. For a long time they were classified as a kingdom...

For lovers of “quiet” hunting, mushroom season begins in early summer and lasts until late autumn. And rarely do they...