Herring under a fur coat - a classic recipe
What would New Year be without champagne, tangerines, Olivier, aspic and everyone’s favorite “Herring under a fur coat”. With the last one...
Side amyotrophic sclerosis(ALS, motor neuron disease, Charcot disease) is a fairly rare pathology of the nervous system in which a person develops muscle weakness and atrophy, which inevitably progresses and leads to fatal outcome. You have already learned about the causes and mechanism of development of the disease from the previous one, let’s now talk about the symptoms, methods of diagnosing and treating ALS.
 Progressive muscle weakness in the legs leads to the patient's inability to move independently.
Progressive muscle weakness in the legs leads to the patient's inability to move independently.
To find out which clinical manifestations may have Charcot's disease, you should understand what central and peripheral motor neurons are.
The central motor neuron is located in the cortex cerebral hemispheres. If it is affected, muscle weakness (paresis) develops in combination with increased muscle tone, reflexes intensify, which are checked with a neurological hammer during examination, pathological symptoms appear (specific reaction of the limbs to certain irritations, for example, extension of the 1st toe when irritating the outer edge of the foot, etc.).
Peripheral motor neuron is located in the brain stem and on various levels spinal cord(cervical, thoracic, lumbosacral), i.e. below the central one. With the degeneration of this motor neuron, muscle weakness also develops, but it is accompanied by decreased reflexes, decreased muscle tone, the absence of pathological symptoms and the development of atrophy of the muscles innervated by this motor neuron.
The central motor neuron transmits impulses to the peripheral one, which transmits impulses to the muscle, and the muscle contracts in response to this. In the case of ALS, at some stage the transmission of the impulse is blocked.
In amyotrophic lateral sclerosis, both central and peripheral motor neurons can be affected, in various combinations and at different levels (for example, there is degeneration of the central motor neuron and peripheral motor neuron at the cervical level or only peripheral motor neuron at the lumbosacral level at the onset of the disease). This is what determines what symptoms the patient will have.
Highlight following forms BASS:
This classification is based on determining the predominant signs of damage to any of the neurons at the onset of the disease. As the disease continues to exist, it loses its significance because pathological process more and more motor neurons are involved at different levels. But such a division plays a role in establishing a diagnosis (is it ALS at all?) and determining the prognosis for life (how long the patient is expected to live).
Common symptoms characteristic of any form of amyotrophic lateral sclerosis are:
With this form of the disease, two options are possible:

Can also debut in two ways:
Amyotrophic lateral sclerosis (ALS) is an incurable, progressive disease of the central nervous system in which the patient experiences damage to the upper and lower motor neurons, which causes muscle atrophy and paralysis. The frequency of this pathology is about 2-7 cases per 100 thousand people. Most often, the disease is diagnosed in patients over 50 years of age.
Scientists have not yet created a unified comprehensive classification of ALS. There are several approaches to classifying the disease. For example, the North American approach involves identifying the following types of ALS: sporadic, familial, sporadic endemic. The classification of amyotrophic lateral sclerosis provides for the following forms of the disease: bulbar, lumbosacral, cervicothoracic and primary generalized. There are also several variants of the disease: mixed, pyramidal and segmental-nuclear.
To the most common initial symptoms diseases include cramps (painful muscle spasms), lethargy and weakness in the distal arms, bulbar disorders, leg muscle atrophy, weakness in shoulder girdle. In addition, different variants of the disease are characterized by different clinical manifestations.
The exact causes of amyotrophic lateral sclerosis are still being investigated by scientists. However, several factors can be named that provoke the disease. For example, about 5% of diseases have a hereditary etiology. At least 20% of cases are associated with mutations in the superoxide dismutase-1 gene. Scientists have proven that important role High activity of the glutamatergic system plays a role in the onset of the disease. The fact is that excess glutamic acid provokes overexcitation and sudden death of neurons. The molecular genetic mechanism of the pathology has also been proven. It is caused by an increase in the level of DNA and RNA in cells, which ultimately leads to disruption of protein synthesis.
Scientists also identify several predisposing factors that play an important role in the occurrence of ALS. First of all, such factors include age. The fact is that the disease usually develops in patients aged 30-50 years. It is worth remembering that only about 5% of patients have hereditary predisposition to BAS. In the vast majority of cases of ALS, the cause of the pathology cannot be determined.
The early course of the disease is characterized by symptoms such as convulsions, twitching, muscle numbness, difficulty speaking, and weakness in the limbs. Since such symptoms are typical for many neurological diseases, diagnose ALS on early stage difficult. In most cases, the disease can be diagnosed at the stage of muscle atrophy.
Depending on the disease affected different parts body, distinguish limb ALS and bulbar ALS. In the first case, patients experience deterioration in ankle flexibility, awkwardness when walking, and they begin to stumble. Bulbar ALS is manifested by difficulty speaking (nasal sound, difficulty swallowing). Soon the patient finds it difficult to move or can no longer move independently. Usually the disease does not have a detrimental effect on the patient’s mental abilities, but it leads to severe depression. In most cases, about three to five years pass from the appearance of the first symptoms to death.
Since ALS is an incurable disease that rapidly shortens a person’s life, the patient’s examination must be comprehensive and accurate. It is extremely important to put correct diagnosis the patient in order to begin timely relief of his main symptoms, as this can prolong the patient’s life. The examination plan usually includes a history of life and illness, a neurological and physical examination, MRI of the spinal cord and brain, EMG, and laboratory tests.
Diagnosis of the disease begins with a detailed interview with the patient. Namely, the doctor needs to clarify whether the patient complains of muscle spasms and twitching, weakness and stiffness, impaired movement of the hands, speech, walking, swallowing, salivation, frequent shortness of breath, weight loss, fatigue, shortness of breath while performing physical exercise. In addition, the doctor should ask whether the patient has noticed double vision, memory loss, crawling sensations on the body, or urinary problems. It is imperative to ask the patient about his family history - whether he has any relatives with chronic movement disorders.
The main purpose of the physical examination is to assess the patient's constitution, weigh him, measure his height, and calculate his body mass index. The neurological examination usually includes neuropsychological testing. When assessing bulbar functions, the doctor pays attention to the timbre of the voice, speed of speech, pharyngeal reflex, the presence of tongue atrophies, and paresis of the soft palate. In addition, during the examination, the strength of the trapezius muscles is checked.
The main instrumental method for diagnosing the disease is needle EMG. This technique allows you to identify signs of the disease such as acute or chronic denervation. In the early stages of the disease, stimulation EMG is ineffective because it does not detect noticeable signs of ALS.
In the process of diagnosing the disease, doctors also use neuroimaging methods. Great importance MRI of the spinal cord and brain plays a role in the differential diagnosis of ALS. During MRI, in 17-67% of patients it is possible to identify symptoms of degeneration of the pyramidal tracts and atrophy of the motor cortex of the brain. However, it is worth noting that this technique is ineffective when diagnosing the disease in patients with bulbar syndrome.
Many laboratory tests are performed during the diagnosis of ALS. In particular, doctors can prescribe clinical and biochemical tests blood, cerebrospinal fluid examination, serological studies. However, the only effective and reliable method of analysis is still considered to be molecular genetic analysis. The presence of mutations in the superoxide dismutase-l gene is considered a suspicion for ALS.
Since the symptoms of amyotrophic lateral sclerosis are similar in many ways to the manifestations of other neurological pathologies, doctors must carry out a differential diagnosis. The most accurate diagnosis can be made using MRI of the brain and spine. First of all, ALS must be differentiated from muscle diseases, which include Rossolimo-Steinert-Kurshman dystrophic myotonia, myositis with cellular abnormalities, and oculopharyngeal myodystrophy.
It is also necessary to distinguish ALS from spinal cord pathologies:
Differential diagnosis is also necessary in order to distinguish the disease from systemic pathologies, lesions of the neuromuscular synapse, and brain pathologies such as multiple system atrophy, dyscirculatory encephalopathy, and syringobulbia.
The main goals of treatment for amyotrophic lateral sclerosis are considered to be to slow the progression of the disease, as well as eliminate its symptoms, which significantly worsen the patient’s quality of life. It should be remembered that ALS is a serious incurable disease that shortens a person’s life expectancy. That is why the doctor has the right to inform the patient of the diagnosis only after a comprehensive and thorough examination.
Treatment of the disease includes drug and non-drug therapy. The latter implies security measures. The patient must limit physical exercise, which may accelerate the progression of ALS. In addition, it is very important to eat properly and nutritiously. Drug therapy is divided into two types: pathogenetic and palliative.
To date, the only drug that can slow the progression of ALS is riluzole. It has been proven that taking it can prolong the patient’s life by an average of three months. This drug is indicated for patients whose disease duration is less than 5 years. The patient should receive 100 mg of the drug daily. To avoid the risk of drug-induced hepatitis, every three months it is necessary to check the levels of AST, ALT and LDH. Since men and smokers have lower concentrations of riluzole in their blood, they should either limit their smoking or quit smoking altogether. bad habit. You will need to take the drug for life.
Scientists have repeatedly tried to use other drugs for pathogenetic therapy. However, such experiments did not prove effective. Among them were:
The effectiveness of taking it has also not been proven high doses Cerebrolysin, despite the fact that this drug is able to slightly improve the condition of patients.
Palliative therapy is intended to eliminate a complex of symptoms of the disease and thereby improve the patient’s quality of life. To eliminate certain symptoms of ALS, the following techniques are used:
To improve muscle metabolism, a patient with ALS can be prescribed the following medications: creatine, carnitine, levocarnitine solution, trimethylhydrazinium propionate. Multivitamin therapy is also indicated for patients, which involves taking multivitamins (neuromultivit, milgamma) and thioctic acid.
In most patients with ALS, the disease is accompanied by serious motor impairments, including limited mobility. Of course, this causes great discomfort to the patient, who constantly needs help from other people. Orthopedic correction techniques help eliminate some movement disorders. The doctor must explain to the patient that the use of assistive devices does not indicate his disability, but only reduces the difficulties caused by the disease.
The most life-threatening symptom of the disease is considered to be respiratory failure. Its earliest symptoms will be morning fatigue, vivid dreams, daytime sleepiness, and dissatisfaction with sleep. For detection respiratory failure At an early stage, polysomnography and spirography are performed. To eliminate apnea, medication and non-invasive ventilation are indicated. It has been proven that these techniques can prolong a patient’s life by one year. If the patient needs assisted breathing for more than 20 hours, the doctor raises the question of a complete transition to invasive ventilation.
Patients who have undergone an initial examination or a repeated diagnosis of the disease must remain under outpatient observation. As any new symptoms appear, they should also receive qualified advice. Patients must take most medications regularly. Only vitamins and myotropic drugs are taken in courses in stages.
Every three months the patient must undergo spirography. If he takes riluzole regularly, he needs to have LDH, AST, and ALT checked every six months. If the patient has dysphagia, blood glucose levels and trophic status should be measured periodically. Patients have a choice of treatment regimen: they can either stay at home or stay in a hospice.
The prognosis for patients with ALS largely depends on the course of the disease. It has been proven that about 80-90% of patients who experience severe respiratory complications die within 3-5 years after the first signs of the disease appear. The remaining 10% of patients have a benign course of the disease. The duration of the disease is significantly reduced in the presence of the following factors: the patient’s age is less than 45 years, bulbar onset of ALS, rapid progression of the disease.
Amyotrophic lateral sclerosis is fatal disease, affecting the nerve cells in the brain and spinal cord that are responsible for movement. The motor neurons (nerve cells that send the impulse to muscle contraction) in an ALS patient die as a result of rapid degeneration, leaving the person paralyzed.
One of famous people British scientist Stephen Hawking has amyotrophic sclerosis.
The muscles controlled by the motor neurons do not have proper nutrition and, as a result, weaken and atrophy (shrink). Paralysis occurs in the final stage of ALS and prevents the patient from functioning physically, although he remains mentally intact. Known causes There are no treatments or treatments for amyotrophic lateral sclerosis, and the disease can affect anyone. The usual cause of death is paralysis of the respiratory muscles that control breathing.
Amyotrophic lateral sclerosis is a progressive disease of the central nervous system.
“A” means “no,” “myo” means muscle cells, and “trophic” refers to nutrition. Nerve cells that extend from the brain to the spinal cord (upper motor neurons) and from the spinal cord to the peripheral nerves (lower motor neurons) inexplicably degenerate and die. "
Lateral" refers to the areas of the spinal cord that are affected, and "sclerosis" occurs when hard fabric replaces previously originally healthy nerve. The parts of the body that are not affected by ALS are those areas that are not involved in the use of motor neurons. The mind remains very sharp and controls vision, hearing, smell, touch and taste. Bowel functions and Bladder, as a rule, are not affected.
Amyotrophic lateral sclerosis rarely causes pain, but patients in the later stages of the disease are dependent on care.
ALS progresses rapidly, and paralyzed patients are usually under intensive care in nursing facilities or loved ones. This can have a devastating psychological impact on family members and the patient. Most cases of ALS are fatal within two to five years, although approximately 10% survive eight years or more.
Amyotrophic lateral sclerosis is not a rare disease. It affects approximately seven people in every 100,000. Most people with ALS are between 40 and 70 years old. Approximately 5-10% of cases show a pattern of heredity.

In 1991, a team of researchers linked familial ALSD to chromosome 21. In 1993, it was discovered that chromosome 21 had structural defects in the SOD1 (superoxide dismutase) gene.
The SOD1 gene is an enzyme that protects motor neurons from free radicals. The island of Guam, Western New Guinea and the Japanese peninsula have high rates of ALS, leading some theorists to believe that the genetic makeup may be susceptible to environmental reasons, such as high levels mercury and lead levels in these areas.

The inheritance pattern is autosomal dominant, meaning that children of an affected parent have a 50% chance of inheriting the disorder. In most cases, it is due to a sporadic gene mutation, which means that the mutation occurs only in the affected person. Sporadic mutations are believed to result from both biological and environmental causes.
IN in rare cases a mutation is evident in NFH, the gene that codes for neurofilament (a structure that maintains cell shape). Familial amyotropic lateral sclerosis has been linked to other chromosomal locations, but the exact genes have not been identified. Thomas Jefferson University in Philadelphia recently approved a project gene therapy BASS.
The project aims to inject an adeno-associated virus carrying a normal copy of the EAAT2 gene into the spinal cord of an ALS patient, where the motor neurons die.

Amyotrophic lateral sclerosis affects everyone, and both men and women are at equal risk. ALS can occur at any age, and the chances of developing it increase as you age. Cases of adolescents with ALS have been reported. A person only needs to inherit a defective gene from one parent to cause the disease.
The disease begins slowly, affecting only one limb, such as an arm or leg, and gradually spreads to more limbs and muscles. When muscles lack the proper nutrition they require, they begin to become thin and deteriorate. This condition is a symptom of amyotrophic lateral sclerosis.
Muscle wasting is caused by the inability of degenerating motor neurons to send signals to the muscles that allow them to function and grow. Typical examples symptoms of ALS are muscle cramps and twitching, weakness in the arms, legs, or ankles, difficulty speaking, and difficulty swallowing. Other early symptoms include stiffness in the arms and legs, loss of a leg, weight loss, fatigue, and difficulty in facial expression.

One of the most early symptoms ALS is weakness in the bulbar muscles. These muscles in the mouth and throat help with chewing, swallowing and speaking. Weakness in these muscle groups typically causes problems such as slurred speech, difficulty speaking, and hoarseness. Another symptom of ALS that usually occurs after the first symptoms appear is persistent muscle twitching (fasciculation).
Fasciculation is almost never the first sign of ALS.
As the disease progresses, the respiratory muscles (breathing muscles) weaken, leading to increased difficulty breathing, coughing, and possibly inhaling food or saliva. The potential for lung infection increases and can lead to death.
Communication becomes very difficult. One way to achieve feedback with other people is to use your eyes.
Blinking is one of the modes that amyotrophic lateral sclerosis patients will be forced to use in order to continue communicating. As the disease progresses, victims gradually lose use of their legs, arms, legs and neck muscles, and paralysis results in the affected muscle groups. They can speak and swallow only with great difficulty.
Sexual function is not affected.
Breathing becomes increasingly difficult, and ALS patients may choose to prolong life with assisted ventilation, which may reduce the risk of death from infections such as pneumonia.

ALS is difficult to diagnose. A number of diagnostic tests will rule in and out others possible reasons and diseases that resemble ALS. Electrodiagnostic tests such as electromyography (EMG) and nerve conduction velocity are used to diagnose ALS.
Blood and urine tests, muscle and/or nerve biopsies are performed, as well as magnetic resonance imaging (MRI), cervical spine myelograms and complete neurological examination Implementation of the program physical therapy, provision wheelchair or walkers, assistance during bathing helps the ALS patient. Other considerations include providing products that are easy to swallow, skin care, and emotional support.
Among neurological diseases there are those that can be successfully treated. But there are also those that can only be suspended - they belong to the group of degenerative-dystrophic ones. One of these diseases is BASS- amyotrophic lateral sclerosis. Read about others here.
At amyotrophic lateral sclerosis gradual death occurs nerve cells, located in the lateral horns of the spinal cord and responsible for motor function.
At the same time, various movement disorders develop. The disease gradually progresses and ultimately leads to fatal outcome due to damage to the respiratory muscles.
ALS is enough rare disease. No more people in the world get it three people out of one hundred thousand. Under the age of 65, the disease is one and a half times more common in men, then men and women are equally affected.
arise amyotrophic lateral sclerosis Can be at any age - from teenagers to old people. In ten percent of cases this family form diseases. The remaining cases are sporadic.
There are several forms of ALS depending on the first part of the spinal cord affected:
There is no single cause for the development of amyotrophic lateral sclerosis; it occurs as a result of a combination of several factors.
Causal factors can come from the body itself and from the external environment:
Symptoms of amyotrophic lateral sclerosis vary depending on the level of damage:
There is a bulbar form - with it the disease begins with the following symptoms:

The most unfavorable variant of the development of amyotrophic lateral sclerosis is the primary generalized form:
ALS - serious illness with the inevitable fatal. Therefore, to make such a diagnosis, it is necessary to conduct a lot of research.
What is taken into account when making a diagnosis:
In this case, paresis and atrophy of the muscles of those parts of the body that are innervated by the affected neurons develop:
Strengthened reflexes, pyramidal signs, and spasticity appear:
A reliable diagnosis of amyotrophic lateral sclerosis is made when peripheral and central motor neurons are damaged in three parts of the spinal cord.
Which instrumental methods carried out to diagnose ALS:
From laboratory methods the only specific thing is genetic mapping— identification of genes responsible for the development of amyotrophic lateral sclerosis.
Differential diagnosis must be carried out in order to exclude all such diseases:
Amyotrophic lateral sclerosis cannot be treated. The only outcome of this disease is death from acute respiratory failure.
However, treatment is still used and has two goals:
To achieve the first goal, only one drug is used - Riluzole. It extends the life of patients with amyotrophic lateral sclerosis by three months.
In most cases, only symptomatic treatment is used:
At posture disorders and foot deformities, corsets and orthopedic shoes are prescribed. For prevention thrombosis — compression stockings or bandaging elastic bandage. If the process is disrupted swallowing- pureed food, feeding through a nasogastric tube.
Update: December 2018
Amyotrophic lateral sclerosis, or Lou Gehrig's disease, is a rapidly progressive disease of the nervous system characterized by damage to motor neurons in the spinal cord, cortex, and brain stem. Also, the motor branches of cranial neurons (trigeminal, facial, glossopharyngeal) are involved in the pathological process.
The disease is extremely rare, approximately 2-5 people per 100,000. It is believed that men over 50 years of age are more often affected. Lou Gehrig's disease makes no exceptions for anyone; it affects people of different social status and various professions (actors, senators, Nobel laureates, engineers, teachers). The most famous patient was world baseball champion Loi Gering, after whom the disease took its name.
In Russia, amyotrophic lateral sclerosis has become widespread. Currently, the number of sick people is approximately 15,000-20,000 in the population. Among the famous people of Russia who have this pathology, we can mention the composer Dmitry Shostakovich, the politician Yuri Gladkov, and the pop singer Vladimir Migulya.
The disease is based on the accumulation of pathological insoluble protein in the motor cells of the nervous system, leading to their death. The cause of the disease is currently unknown, but many theories exist. The main theories include:
Any form of the disease has the same onset: patients complain of increasing muscle weakness, decrease in muscle mass and the appearance of fasciculations (muscle twitching).
Bulbar form of ALS
characterized by symptoms of damage to the cranial nerves (9, 10 and 12 pairs):Cervicothoracic variant The disease primarily affects the patient’s upper limbs, symmetrically on both sides:
Lumbosacral form usually begins with a feeling of weakness in the lower extremities.
Regardless of which variant predominates in patients at the beginning of the disease, the outcome is still the same. The disease progresses steadily, spreading to all muscles of the body, including the respiratory muscles. When the respiratory muscles fail, the patient begins to need artificial ventilation lungs and constant care.
In my practice, I observed two patients with ALS, a man and a woman. They were distinguished by their red hair color and relatively young age (up to 40 years). Outwardly, they were very similar: there was no hint of muscles, an amicable face, and their mouth was always slightly open.
Such patients die in most cases from concomitant diseases(pneumonia, sepsis). Even with proper care, they develop bedsores (see), hypostatic pneumonia. Realizing the severity of their illness, patients fall into depression, apathy, and cease to be interested in the outside world and their loved ones.
Over time, the patient's psyche undergoes strong changes. The patient I observed for a year was distinguished by his capriciousness, emotional lability, aggressiveness, and lack of restraint. Intelligence tests showed a decline in his thinking, mental abilities, memory, attention.
The main diagnostic methods include:
What happens in ALS
Due to the fact that ALS has similar symptoms to other diseases, differential diagnosis is made:
Treatment of the disease is currently ineffective. Medications and proper care for the patient only extend life expectancy without providing full recovery. Symptomatic therapy includes:
Complex therapy necessarily includes feeding through a nasogastric tube, massage, exercise therapy with a doctor, and consultations with a psychologist.
Sadly, the prognosis for amyotrophic lateral sclerosis is unfavorable. Patients die literally within a few months or years, average duration life in patients:
A more favorable prognosis for hereditary cases of the disease associated with mutations in the superoxide dismutase-1 gene.
The situation in Russia is overshadowed by the fact that patients are not provided with proper care, as evidenced by the fact that Riluzote, a drug that slows the course of the disease, was not even registered in Russia until 2011, and only in the same year the disease itself was included in the list " rare." But in Moscow there are:
Finally, I would like to add about the Ice Bucket Challenge charity event that took place in July 2014. It was aimed at raising funds to support patients with amyotrophic lateral sclerosis and became quite widespread. The organizers managed to raise more than $40 million.
The essence of the action was that a person either doused himself with a bucket of ice water and captured it on video, or donated a certain amount of money to a charitable organization. The event became quite popular due to the participation of popular performers, actors and even politicians.

What would New Year be without champagne, tangerines, Olivier, aspic and everyone’s favorite “Herring under a fur coat”. With the last one...

Let's prepare the necessary ingredients for the cookies. The first thing to do is put the water to boil. We need...

Is it possible to register an employee for the position of financial director - chief accountant? The chief accountant claims that yes, but...

The head of a small business can easily manage the budget independently. CHECKED! If you're budgeting...

The creation of new projects involves a preliminary economic justification for their feasibility, subsequent...
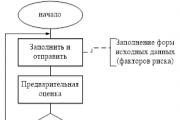
Reporting is generated by the RM, is agreed upon (approved) by the Risk Committee under the Management Board and transmitted to...

At the edge of a large, very large meadow, on a long emerald blade of grass lived a tiny Ladybug. God's little...

Nowadays, it is quite common for people to turn to the stars. With the help of a horoscope a person can find out...

Business or friendly. If you were pursuing a stranger, it means your level of trust in the female sex...

according to Freud's dream book If you dreamed about how you were fishing, it means that in real life you can hardly switch off...

The New Year's whirlwind will spin us around so quickly and rapidly that it's time to thoroughly think through the New Year's...

The article offers you some of the most delicious recipes for making vinaigrette. Vinaigrette is a delicious salad...

July 4th, 2015 , 08:33 pm Lately our Nastena has been completely giving up sweets (and thank God), but...

Fragrant, very tasty chocolate cake, it’s impossible to stop eating! The Negro’s Kiss cake is very easy to prepare,...

Let's prepare the necessary ingredients for the cookies. The first thing to do is put the water to boil. Us...

Is it possible to register an employee for the position of financial director - chief accountant? The chief accountant claims...