Visão filosófica do problema
O sentido da vida está relacionado com a questão “Para que viver”, e não com a questão de como manter a vida. A atitude de uma pessoa é...
O fenômeno associado à destruição dos glóbulos vermelhos e à liberação de hemoglobina no plasma é a hemólise. Existem várias classificações deste processo, dependendo das causas que o provocam, do local de origem, etc.
Nem todas as pessoas sabem o que é e se representa um perigo. O processo ocorre no corpo depois que os glóbulos vermelhos cumpriram seu prazo - 4-5 meses. No final disso, as células morrem.
O perigo é a destruição dos glóbulos vermelhos em ritmo acelerado, pois existe o risco de desenvolver patologias.
Tipos de hemólise:
.jpg)
No primeiro caso, as células que cumpriram o prazo são substituídas por novas, e o processo é dividido em:
A destruição patológica é a morte de glóbulos vermelhos viáveis sob qualquer influência. O processo é classificado de acordo com os fatores de impacto:


Também se destaca a hemólise elétrica - a morte dos glóbulos vermelhos devido à exposição à corrente elétrica.
A degradação dos glóbulos vermelhos ocorre por vários motivos. Na hemólise aguda, observa-se um curso acelerado da reação e uma deterioração significativa na condição da pessoa.
As principais razões que contribuem para isso:
O aparecimento de hemólise patológica é causado por:
A anemia falciforme e o tratamento descontrolado com medicamentos podem causar danos ao sangue. Alguns analgésicos, sulfonamidas, diuréticos e medicamentos para o tratamento da tuberculose podem provocar aumento da morte de glóbulos vermelhos.

A hemólise é possível devido a violações durante os testes, o que os torna inadequados para pesquisas futuras. Isso acontece como resultado de coleta de sangue muito rápida, falha na manutenção da esterilidade, armazenamento e transporte inadequados, o que causa destruição das membranas.
O despreparo do paciente para a análise, por exemplo, ingerir alimentos muito gordurosos no dia anterior, também impacta negativamente, pois a decomposição das gorduras estimula o desenvolvimento de hemólise.
É detectada imediatamente ao nascimento e é causada pela incompatibilidade dos anticorpos da mãe e do filho. Em crianças, inchaço intenso, anemia e icterícia são pronunciados. Assim como nos adultos, a patologia é dividida em intravascular e intracelular.
A incompatibilidade entre o sangue do feto e da mãe é determinada durante a gravidez e muitas vezes é tratada no útero. Na maioria das vezes, um bebê nasce cesariana. Doença hemolítica geralmente ocorre em bebês prematuros.
.jpg)
O tratamento posterior da criança e, às vezes, da mãe, é realizado com base no quadro clínico. Inclui transfusões de sangue e terapia hormonal.
Simultaneamente à transfusão de sangue, é realizado o tratamento com glicocorticosteroides, por exemplo, com o medicamento Cortisona, administrado por via intramuscular.
Muitas vezes você tem que se recusar a alimentar seu bebê leite materno, que se refere a métodos de tratamento não medicamentosos.
Para uma pessoa saudável, a hemólise biológica dos glóbulos vermelhos passa despercebida. Sintomas clínicos possível com manifestações agudas ou patológicas.
Sinais de hemólise aguda:
Se o paciente estiver consciente, pode haver queixas sobre:
.jpg)
Não há sintomas claros que indiquem que os glóbulos vermelhos estejam se desintegrando em pessoas submetidas a terapia hormonal, radioterapia e sob anestesia, ou que sua manifestação seja insignificante.
A análise laboratorial das amostras colhidas mostra claramente que os glóbulos vermelhos estão se decompondo, a reação sanguínea mostra que a anemia está aumentando, as plaquetas estão diminuindo, a bilirrubina está aumentando e a coagulação está prejudicada.
A cor da urina também muda, fica vermelha escura, a análise bioquímica mostra a presença de hemoglobina, potássio e proteínas.
Para determinar a hemólise, são medidos o nível de hemoglobina, o número de reticulócitos e a bilirrubina no soro. Ocasionalmente, é necessário medir o ciclo de vida dos glóbulos vermelhos utilizando métodos de radioisótopos.
Para determinar se a degradação dos glóbulos vermelhos é normal, é necessário determinar a densidade de sua membrana pelo método da resistência osmótica, que permite identificar a destruição mínima ou máxima.

Após a coleta de sangue, é realizado um teste especial - o índice de hemólise (HI), que permite determinar o teor de hemoglobina no sangue. Nos homens, o conteúdo ideal de eritrócitos é 4,3-5,7*106/μl, nas mulheres - 3,9-5,3*106/μl. O número de glóbulos vermelhos em uma criança menor de 12 anos é 3,6-4,9*1012/l, 12-15 anos - 3,9-5,5*1012/l.

Além disso, em estudos laboratoriais, é determinado como a razão entre o volume total de glóbulos vermelhos e o volume total de plasma.
O valor ideal para homens e mulheres é 0,4-0,52 e 0,37-0,49, respectivamente.
A norma do hematócrito em crianças do primeiro dia de vida até um mês é de 0,56 a 0,45, de um ano a 15 anos - 0,35-0,39, maiores de 15 anos - 0,47.
Determinar a esfericidade dos glóbulos vermelhos não é de pouca importância. Esta é a relação entre o diâmetro e a espessura da parede. Normalmente, o valor em humanos é 0,26-0,28.
Os glóbulos vermelhos que cumpriram sua vida têm uma forma esférica. Se uma configuração semelhante for observada nas células jovens, sua vida útil será reduzida em 10 vezes e elas morrerão sem cumprir sua função.
O aparecimento de células sanguíneas esféricas permite tirar conclusões sobre um aumento no índice de esfericidade, o que indica o desenvolvimento de anemia hemolítica.
As células mais viáveis são as células jovens (), que acabaram de emergir de medula óssea. Devido ao seu formato em forma de disco espessado, eles têm índice baixo esfericidade.
Se a análise revelar aumento da degradação dos glóbulos vermelhos, é prescrita uma nova doação de sangue para eliminar erros durante a amostragem e garantir a confiabilidade do resultado.
A hemólise aguda requer cuidados médicos de emergência. Parar as manifestações de uma crise só é possível em condições de internação, na unidade de terapia intensiva.
Os principais métodos de tratamento incluem:
O tratamento não é fácil anemia hereditária natureza hemolítica. Às vezes é necessário remover o baço, especialmente se houver danos extensos ao órgão.
.jpg)
Freqüentemente, é necessário um procedimento de purificação do sangue por meio de plasmaférese com o uso (por via intravenosa) do medicamento Heparina, que ajuda a remover a hemoglobina livre.
Para sinais de hemólise autoimune, são utilizados glicocorticosteróides, por exemplo Prednisolona. Uma crise hemolítica no estágio profundo pode ser interrompida com a ajuda de Reogluman.
Uma medida preventiva para insuficiência renal é o uso combinado de Diacarb e bicarbonato de sódio.
Com a hemólise, a principal consequência é a anemia hemolítica, muitas vezes acompanhada de alteração no número de plaquetas, leucócitos, desenvolvimento de coágulos sanguíneos nos vasos e ocorrência de colelitíase.
Para fins de prevenção, é necessário seguir regras simples:
É importante não se automedicar escolhendo medicamentos aleatórios. Somente um especialista pode prescrever a terapia necessária, com base em exames e exames.
A doença hemolítica do recém-nascido (HDN) é uma doença muito comum. Esta patologia é registrada em aproximadamente 0,6% das crianças nascidas. Apesar do desenvolvimento vários métodos tratamento, a taxa de mortalidade por esta doença chega a 2,5%. Infelizmente, há um grande número de “mitos” cientificamente infundados sobre esta patologia. Para uma compreensão profunda dos processos que ocorrem durante a doença hemolítica, o conhecimento do normal e fisiologia patológica, e também, claro, obstetrícia.

O CTT é consequência de um conflito entre o sistema imunológico da mãe e do filho. A doença se desenvolve devido à incompatibilidade do sangue de uma mulher grávida com antígenos na superfície dos glóbulos vermelhos fetais (principalmente estes). Simplificando, eles contêm proteínas que são reconhecidas pelo corpo da mãe como estranhas. É por isso que os processos de ativação do sistema imunológico começam no corpo da gestante. O que está acontecendo? Assim, em resposta à entrada de uma proteína desconhecida, ocorre a biossíntese de moléculas específicas que podem entrar em contato com o antígeno e “neutralizá-lo”. Essas moléculas são chamadas de anticorpos, e a combinação de anticorpo e antígeno é chamada de imunocomplexos.

Porém, para chegar um pouco mais perto de uma verdadeira compreensão da definição de HDN, é necessário compreender o sistema sanguíneo humano. Há muito se sabe que o sangue contém tipos diferentes células. O maior número da composição celular é representado pelos eritrócitos. Sobre nível moderno No desenvolvimento da medicina, são conhecidos pelo menos 100 sistemas diferentes de proteínas antigênicas presentes na membrana dos eritrócitos. Os mais estudados são os seguintes: rhesus, Kell, Duffy. Mas, infelizmente, existe um equívoco muito comum de que a doença hemolítica do feto se desenvolve apenas de acordo com o grupo ou antígenos Rh.
A falta de conhecimento acumulado sobre as proteínas da membrana eritrocitária não significa que a incompatibilidade com este antígeno específico em uma mulher grávida seja excluída. Este é o desmascaramento do primeiro e, talvez, o mais básico mito sobre as causas desta doença.
Fatores que causam conflito imunológico:

Muitas vezes, uma mulher com Rh negativo se preocupa com seus futuros filhos, mesmo sem estar grávida. Ela tem medo da possibilidade de desenvolver um conflito Rhesus. Alguns têm até medo de se casar com um homem Rh positivo.

Mas isso é justificado? E qual é a probabilidade de desenvolver um conflito imunológico nesse casal?
Felizmente, o sinal Rh é codificado pelo chamado genes alélicos. O que isso significa? O fato é que as informações localizadas nas mesmas áreas dos cromossomos emparelhados podem ser diferentes:
O resultado final é que uma pessoa Rh positiva pode conter dois traços dominantes (RR) ou dominantes e recessivos (Rr) em seus cromossomos.
Além disso, uma mãe Rh negativa contém apenas dois traços recessivos (rr). Como você sabe, durante a herança, cada pai pode dar apenas uma característica ao filho.
| Mãe (r) (r) | Pai (R) (R) | |
|---|---|---|
| Criança | (R)+(r) Rh positivo | (R)+(r) Rh positivo |
| Probabilidade | 100% | 100% |
Assim, em 50% dos casos, pode não haver nenhum conflito imunológico se o pai for portador do traço recessivo do fator Rh.
Assim, podemos tirar uma conclusão simples e óbvia: o julgamento de que uma mãe Rh negativo e um pai Rh positivo devem necessariamente ter incompatibilidade imunológica é fundamentalmente errado. Esta é a “exposição” do segundo mito sobre as causas do desenvolvimento da doença hemolítica do feto.
Além disso, mesmo que a criança ainda apresente fator Rh positivo, isso não significa que o desenvolvimento de cefaleia do tipo tensional seja inevitável. Não se esqueça propriedades protetoras. Durante uma gravidez fisiológica, a placenta praticamente não permite a passagem de anticorpos de mãe para filho. Prova disso é o fato de que a doença hemolítica ocorre apenas no feto de cada 20 mulheres Rh negativo.

Ao saber da identidade de seu sangue, mulheres com uma combinação semelhante de grupo e Rhesus entram em pânico. Mas quão justificados são esses medos?
À primeira vista, pode parecer que a combinação de “dois males” criará um alto risco de desenvolvimento de CTT. Contudo, a lógica comum não funciona aqui. É o contrário: a combinação desses fatores, curiosamente, melhora o prognóstico. E há uma explicação para isso. No sangue de uma mulher com o primeiro grupo sanguíneo já existem anticorpos que reconhecem uma proteína estranha nos glóbulos vermelhos de um grupo diferente. Assim foi pretendido pela natureza, esses anticorpos são chamados de aglutininas alfa e beta, todos os representantes do primeiro grupo os possuem. E quando atingido grande quantidade glóbulos vermelhos fetais na corrente sanguínea da mãe, eles são destruídos pelas aglutininas existentes. Assim, os anticorpos para o sistema do fator Rh simplesmente não têm tempo de se formar, porque as aglutininas estão à frente deles.
Mulheres com o primeiro grupo e Rh negativo apresentam um pequeno título de anticorpos contra o sistema Rh e, portanto, a doença hemolítica se desenvolve com muito menos frequência.
Não repitamos que Rh negativo ou primeiro grupo sanguíneo já é um certo risco. No entanto, É importante saber da existência de outros fatores predisponentes:

Isto é especialmente verdadeiro para aqueles que tiveram várias reações alérgicas após uma transfusão. Muitas vezes, na literatura, pode-se encontrar a opinião de que estão em risco aquelas mulheres que receberam transfusão de tipo sanguíneo sem levar em conta o fator Rh. Mas isso é possível em nosso tempo? Esta possibilidade está praticamente excluída, uma vez que o estado de Rhesus é verificado em várias etapas:
Surge a questão: onde então é possível que uma mulher fique sensibilizada (presença hipersensibilidade e anticorpos) aos glóbulos vermelhos Rh-positivos?
A resposta foi dada recentemente, quando os cientistas descobriram que existe um grupo dos chamados “doadores perigosos”, cujo sangue contém glóbulos vermelhos com um antígeno Rh positivo fracamente expresso. É por esta razão que seu grupo é definido pelos laboratórios como Rh negativo. Porém, quando esse sangue é transfundido, o corpo do receptor pode começar a produzir anticorpos específicos em pequeno volume, mas mesmo sua quantidade é suficiente para que o sistema imunológico “lembre” desse antígeno. Portanto, em mulheres com situação semelhante, mesmo no caso da primeira gravidez, pode surgir um conflito imunológico entre o corpo dela e o filho.
Acredita-se que em Durante a primeira gravidez, o risco de desenvolver um conflito imunológico é mínimo. E a segunda gravidez e as subsequentes já ocorrem com formação de anticorpos e incompatibilidade imunológica. E de fato é. Mas muitas pessoas esquecem que a primeira gravidez deve ser considerada o fato do desenvolvimento do óvulo fertilizado no corpo da mãe a qualquer prazo.
Portanto, mulheres que tiveram:
Além disso, primigestas com as seguintes patologias também apresentam risco aumentado:
Obviamente, a primeira gravidez nem sempre significa ausência de complicações e desenvolvimento de conflito imunológico. Este facto dissipa o mito de que apenas a segunda gravidez e as subsequentes são potencialmente perigosas.
Não há diferenças fundamentais nesses conceitos. Simplesmente a doença hemolítica no feto ocorre no período pré-natal. HDN significa a ocorrência de um processo patológico após o nascimento de um filho. Por isso, a diferença está apenas nas condições em que o bebê se encontra: no útero ou após o nascimento.
Mas há mais uma diferença no mecanismo desta patologia: durante a gravidez, os anticorpos maternos continuam a entrar no corpo do feto, o que leva a uma deterioração do estado do feto, enquanto após o parto este processo é interrompido. Por isso mulheres que deram à luz um bebê com doença hemolítica estão estritamente proibidas de alimentar seu bebê com leite materno. Isto é necessário para evitar a entrada de anticorpos no corpo do bebê e não agravar o curso da doença.

Existe uma classificação que reflete bem as principais formas da doença hemolítica:
1. Anêmico– o principal sintoma é a diminuição do feto, que está associada à destruição dos glóbulos vermelhos () no corpo do bebê. Essa criança tem todos os sinais:

2. Forma de edema. O sintoma predominante é a presença de edema. Característica distintivaé a deposição de excesso de líquido em todos os tecidos:
3. Forma de icterícia caracterizado por, que é formado como resultado da destruição dos glóbulos vermelhos. Esta doença causa danos tóxicos a todos os órgãos e tecidos: 
4. Combinado (o mais grave) – é uma combinação de todos os sintomas anteriores. É por esta razão que este tipo de doença hemolítica apresenta a maior taxa de mortalidade.
Para avaliar corretamente a condição da criança e, o mais importante, prescrever um tratamento eficaz, é necessário utilizar critérios confiáveis na avaliação do grau de gravidade.
Já durante a gravidez é possível determinar não só a presença da doença, mas até a gravidade.

Os métodos mais comuns são:
1. Determinação do título de anticorpos Rh ou de grupo. Acredita-se que um título de 1:2 ou 1:4 não seja perigoso. Mas esta abordagem não se justifica em todas as situações. Aqui reside outro mito de que “quanto maior o título, pior o prognóstico”.
O título de anticorpos nem sempre reflete a real gravidade da doença. Ou seja, este indicador é muito relativo. Portanto, é necessário avaliar a condição do feto por meio de diversos métodos de pesquisa.
2. O diagnóstico por ultrassom é um método muito informativo. Os sinais mais característicos:
3. Aumento da densidade do líquido amniótico.
4. No momento do registro - sinais e distúrbios do ritmo cardíaco.
5.B em casos raros realizar pesquisas Sangue do cordão umbilical (determinar o nível de hemoglobina e bilirrubina). Este método é perigoso devido à interrupção prematura da gravidez e morte fetal.
6. Após o nascimento de uma criança, existem métodos de diagnóstico mais simples:

O tratamento para esta doença pode começar agora. durante a gravidez, para evitar a deterioração do estado do feto:
Métodos de tratamento de uma criança após o parto:

Para doenças graves, são utilizados os seguintes métodos de tratamento:

Mulheres em risco de desenvolver incompatibilidade imunológica deve ser respeitado seguindo regras, existem apenas dois deles:
A doença hemolítica do recém-nascido é uma doença grave e muito perigosa.
No entanto, não se deve acreditar incondicionalmente em todos os “mitos” sobre esta patologia, embora alguns deles já estejam firmemente arraigados entre a maioria das pessoas. Uma abordagem competente e uma validade científica rigorosa são a chave para uma gravidez bem sucedida. Além disso, é necessário dar a devida atenção às questões de prevenção para evitar ao máximo possíveis problemas.Hemólise- trata-se da destruição fisiológica das células sanguíneas, nomeadamente das células da série eritrocitária, reflectindo o processo natural do seu envelhecimento. A destruição direta das células sanguíneas eritrocitárias ocorre sob a influência da hemolisina, que na maioria das vezes desempenha o papel de toxinas bacterianas.
Dependendo da origem, todas as variantes do curso da reação hemolítica podem ser atribuídas a uma das duas opções principais: natural ou patológica. A hemólise natural é uma cadeia contínua de processos químicos, como resultado da “renovação fisiológica” da composição dos glóbulos vermelhos, desde que funcionamento normal estruturas do sistema reticuloendotelial.
Variantes de reações hemolíticas observadas em condições laboratoriais incluem temperatura e hemólise osmótica. No primeiro tipo de hemólise, uma cadeia de reações hemolíticas é desencadeada como resultado da exposição a baixas temperaturas críticas nos componentes sanguíneos. Na hemólise osmótica, os glóbulos vermelhos são destruídos quando o sangue entra em um ambiente hipotônico. Para pessoas saudáveis caracterizado pela resistência osmótica mínima dos eritrócitos, que está dentro de 0,48% de NaCl, enquanto a destruição completa da maior parte dos eritrócitos é observada em uma concentração de NaCl de 0,30%.
Numa situação em que o paciente apresenta endotoxemia causada pela ação de microrganismos infecciosos, criam-se condições para o desenvolvimento de hemólise biológica. Uma reação hemolítica semelhante é observada durante a transfusão de substâncias incompatíveis cheio de sangue ou seus componentes.
Outra variante da reação hemolítica é o tipo mecânico de hemólise, cujo aparecimento de sintomas é facilitado pelo fornecimento de impacto mecânico ao sangue (por exemplo, agitando um tubo contendo sangue). Esse tipo de reação hemolítica é típica de pacientes submetidos à troca valvar cardíaca.
Existe toda uma gama de substâncias com propriedades hemolisantes ativas, entre as quais as mais ativas são: venenos de cobra e veneno de inseto. O desenvolvimento da hemólise é facilitado pela exposição a uma série de produtos químicos do grupo do clorofórmio, da gasolina e até do álcool.
Uma forma etiopatogenética rara e ao mesmo tempo mais grave de reação hemolítica para o paciente é a hemólise autoimune, cuja ocorrência é possível se o corpo do paciente produzir anticorpos contra seus próprios glóbulos vermelhos. Esta patologiaé acompanhada por anemia grave do corpo e liberação de hemoglobina na urina em uma concentração criticamente alta.
Numa situação em que a pessoa não apresenta sinais de hemólise patológica e a destruição das hemácias ocorre conforme planejado com a participação das estruturas do sistema reticuloendotelial de acordo com o tipo intracelular, não manifestações externas uma pessoa não sentirá hemólise.
O quadro clínico de hemólise é observado apenas no caso de curso patológico e inclui vários períodos: crise hemolítica ou hemólise aguda, fase subcompensada de hemólise e período de remissão.
O desenvolvimento de hemólise aguda, caracterizada por um curso extremamente rápido que piora significativamente a saúde do paciente, é mais frequentemente observado com transfusão de hemocomponentes incompatíveis, graves lesão infecciosa efeitos corporais e tóxicos, por exemplo, tomar medicamentos. O perigo desta condição é que a reação hemolítica é tão intensa que o corpo não possui capacidade compensatória para produzir um número suficiente de glóbulos vermelhos. Nesse sentido, os sintomas clínicos de uma crise hemolítica consistem em manifestações de intoxicação por bilirrubina e uma forma grave de síndrome anêmica. Os sinais específicos de crise hemolítica aguda que ocorre no intraoperatório são o aparecimento de sangramento excessivo desmotivado na superfície da ferida, bem como a liberação de urina escura pelo cateter.
As manifestações de intoxicação por bilirrubina são descoloração pele na forma de icterícia, de natureza difusa e intensa. Além disso, o paciente sente náuseas intensas e vômito repetido dor que não tem relação com a ingestão de alimentos, dor intensa na cavidade abdominal, que não tem localização clara. Na crise hemolítica grave, o paciente evolui rapidamente síndrome convulsiva e vários graus de comprometimento da consciência.
Os sintomas que refletem a síndrome anêmica são fraqueza grave e incapacidade de realizar atividades normais. atividade física, palidez visual da pele, distúrbios respiratórios na forma de falta de ar crescente e um exame objetivo do paciente muitas vezes revela sopro sistólico na projeção da ausculta do ápice do coração. Um sintoma patognomônico da hemólise patológica intracelular é um aumento no tamanho do baço e do fígado, e a hemólise intravascular é caracterizada por uma alteração na urina na forma de escurecimento.
Um reflexo específico da hemólise aguda é o aparecimento de alterações específicas nos exames de sangue e urina na forma de bilirrubinemia e hemoglobinemia graves, e diminuição dos fatores de fibrinólise, hemoglobinúria e aumento significativo da creatinina e uréia.
O perigo de hemólise ocorrer de forma aguda é possível desenvolvimento complicações na forma de crise de regeneração e insuficiência renal aguda.
Na fase subcompensatória da hemólise, os processos de produção de células sanguíneas pelo broto eritróide da medula óssea são ativados, portanto a gravidade das manifestações clínicas diminui, mas permanecem manifestações cutâneas e hepatoesplenomegalia. A síndrome anêmica nesta fase da hemólise praticamente não é observada, e os exames clínicos de sangue mostram número aumentado reticulócitos, refletindo o processo regenerativo no sangue.
Uma forma especial de reação hemolítica é a doença hemolítica, observada em crianças durante o período neonatal. Ainda no período pré-natal, o feto apresenta manifestações hemolíticas causadas por incompatibilidade hemograma mãe e feto. A intensidade do desenvolvimento da reação hemólise tem uma clara correlação com a magnitude do aumento do título de anticorpos no sangue de uma mulher grávida.
A apresentação clínica da hemólise em recém-nascidos pode ocorrer de três formas clássicas. A opção mais desfavorável para a recuperação da criança é a variante edemaciada, que aumenta significativamente o risco de natimorto. Além do inchaço pronunciado dos tecidos moles, há acúmulo excessivo de líquido nas cavidades naturais (pleural, pericárdica, cavidade abdominal).
A síndrome da icterícia se manifesta por alterações na cor da pele, líquido amniótico e lubrificação do vérnix. A criança apresenta sinais de danos tóxicos nas estruturas do centro sistema nervoso na forma de aumento da prontidão convulsiva, rigidez e opistótono, distúrbios oculomotores e sintoma do “sol poente”. O aparecimento destes sintomas pode ser fatal.
A síndrome anêmica no recém-nascido, via de regra, não é acompanhada de manifestações clínicas pronunciadas e consiste apenas em alterações nos exames laboratoriais. A duração da síndrome anêmica com curso favorável de hemólise em um recém-nascido, via de regra, não ultrapassa três meses.
Forneceu o funcionamento normal de todos os órgãos e sistemas corpo humano, os processos de formação dos glóbulos vermelhos e sua destruição estão em equilíbrio. A localização predominante do processo de destruição das células sanguíneas eritrocitárias são as estruturas do sistema reticuloendotelial, cujos principais representantes são o baço e o fígado, onde se observa a fragmentação do eritrócito e sua posterior lise. À medida que os glóbulos vermelhos envelhecem, perdem a elasticidade e a capacidade de mudar de forma, dificultando a sua passagem pelos seios esplênicos. O resultado desse processo é a retenção de glóbulos vermelhos no baço e seu posterior sequestro.
Na verdade, nem todos os glóbulos vermelhos que circulam na corrente sanguínea passam pelos seios esplênicos, mas apenas 10% de sua massa total. Devido ao fato de as fenestras dos seios vasculares terem um lúmen significativamente menor que o diâmetro de um glóbulo vermelho padrão, as células velhas, caracterizadas pela rigidez da membrana, ficam retidas nos sinusóides. Posteriormente, os glóbulos vermelhos sofrem distúrbios metabólicos, causada por baixa acidez e baixa concentração de glicose na região dos seios esplênicos. A eliminação dos glóbulos vermelhos retidos nos seios da face ocorre com a ajuda de células macrófagas que estão constantemente presentes no baço. Assim, a hemólise intracelular é a destruição direta das células sanguíneas da série eritrocitária pelos macrófagos do sistema reticuloendotelial.
Dependendo da localização predominante do processo de destruição dos glóbulos vermelhos, distinguem-se duas formas principais: hemólise intracelular e intravascular.
A hemólise extravascular destrói até 90% dos glóbulos vermelhos, desde que as estruturas do sistema reticuloendotelial estejam funcionando normalmente. A destruição da hemoglobina consiste na clivagem primária das moléculas de ferro e globina e na formação de biliverdina sob a influência da heme oxigenase. Posteriormente, é lançada uma cadeia de reações enzimáticas, cujo produto final é a formação da bilirrubina e sua entrada na corrente sanguínea geral. Nesta fase, são ativados os hepatócitos, cuja função visa absorver a bilirrubina do plasma sanguíneo. Numa situação em que o paciente apresenta um aumento significativo na concentração de bilirrubina no sangue, parte dela não se liga à albumina e é filtrada nos rins.
A adsorção da bilirrubina do plasma ocorre no parênquima hepático pela ativação das estruturas do sistema de transporte, após o que é conjugada com o ácido glucurônico. Esta transformação química ocorre com a participação de um grande número de catalisadores enzimáticos, cuja atividade depende diretamente do estado dos hepatócitos. Uma criança recém-nascida tem baixa atividade enzimática fígado e, portanto, a hemólise excessiva em crianças é causada precisamente pela incapacidade do fígado de conjugar a bilirrubina com rapidez suficiente.
A transformação adicional da hemoglobina conjugada envolve sua liberação pelos hepatócitos juntamente com a bile, que também contém outros complexos (fosfolipídios, colesterol, sais biliares). Na luz das vias biliares, a bilirrubina sofre uma cadeia de alterações sob a influência da enzima desidrogenase e da formação do urobilinogênio, que é absorvido pelas estruturas duodeno e sofre oxidação adicional no fígado. Parte da bilirrubina que não é absorvida no intestino delgado entra no intestino delgado, onde uma nova forma é formada - o estercobilinogênio.
A maior parte do estercobilinogênio é excretada nas fezes e o restante é excretado na urina como urobilina. Assim, a hemólise intensiva dos eritrócitos pode ser monitorada pela determinação da concentração de estercolibina. Ao mesmo tempo, para avaliar a intensidade da hemólise, não se deve considerar o aumento da concentração de urobilinogênio, que aumenta não só na situação de aumento da hemólise, mas também com dano morfológico e funcional à massa de hepatócitos.
Os principais critérios diagnósticos que refletem o processo de aumento da hemólise intracelular são o aumento da concentração da fração conjugada da bilirrubina, bem como o aumento acentuado da liberação de estercobilina e urobilina com fluidos biológicos naturais. O desenvolvimento de hemólise intracelular patológica é facilitado pela deficiência hereditária da membrana eritrocitária do paciente, produção prejudicada de hemoglobina, bem como um número excessivo de células sanguíneas eritrocitárias, que ocorre com icterícia fisiológica.
Na hemólise intravascular fisiológica, a destruição das células sanguíneas eritrocitárias ocorre diretamente na corrente sanguínea circulante, e o componente desse tipo de reação hemolítica não excede 10% da massa total de eritrócitos destruídos. A reação normal de hemólise intravascular é acompanhada pela liberação de hemoglobina e pela ligação desta às globulinas plasmáticas. O complexo resultante entra nas estruturas do sistema reticuloendotelial e sofre novas transformações.
A hemólise intravascular maciça é acompanhada por uma capacidade reduzida de ligação à hemoglobina das globulinas plasmáticas, o que se reflete na liberação de grandes quantidades de hemoglobina através das estruturas do trato urinário. Ao entrar nos rins, a hemoglobina provoca alterações em suas estruturas na forma de deposição de hemossiderina na superfície do epitélio dos túbulos renais, o que provoca diminuição da reabsorção tubular e liberação de hemoglobina livre junto com a urina.
Deve-se ter em mente que não existe uma correlação clara entre o grau de hemoglobinemia e a intensidade da excreção de hemoglobina livre na urina. Assim, uma capacidade reduzida de ligação à hemoglobina do plasma é acompanhada pelo desenvolvimento de hemoglobinúria, mesmo com um ligeiro aumento na concentração de hemoglobina no sangue. Assim, os principais marcadores de aumento da hemólise intravascular são o aumento da concentração de bilirrubina livre na urina e no sangue, bem como a hemossiderinúria concomitante.
Devido ao fato de a crise hemolítica aguda pertencer à categoria de condições de emergência, especialistas desenvolveram um algoritmo unificado para fornecer assistência emergencial para esta categoria de pacientes, incluindo uma componente medicinal e não medicinal. Alívio dos sinais de crise hemolítica em período agudo deve ser realizada somente em hospital hematológico em leitos de unidade de terapia intensiva.
Numa situação em que a hemólise é acompanhada declínio crítico nível de hemoglobina, o único método eficaz de tratamento é a transfusão de glóbulos vermelhos num volume diário estimado de 10 ml por 1 kg de peso corporal do paciente. Em caso de sinais existentes de crise regenerativa, recomenda-se complementar a terapia transfusional esteróides anabolizantes(Retabolil na dose de 25 mg uma vez a cada 2 semanas).
A presença de sinais de hemólise autoimune aguda em um paciente é a base para o uso de glicocorticosteroides. A dose diária inicial de Prednisolona é de 60 mg, mas em algumas situações a dose pode ser aumentada para 150 mg. Depois de parar a crise, é aconselhável reduzir gradualmente a dosagem (não mais que 5 mg por dia) até um nível de 30 mg. A redução adicional da dosagem envolve tomar o medicamento com uma dose menor de 2,5 mg a cada cinco dias até a retirada completa.
Numa situação em que a terapia com glicocorticosteróides não surte o efeito desejado na forma de períodos de remissão de 7 meses ou mais, recomenda-se que o paciente seja submetido à remoção cirúrgica do baço.
As formas refratárias de hemólise autoimune envolvem o uso simultâneo de medicamentos glicocorticosteróides e medicamentos imunossupressores (Imuran na dose diária calculada de 1,5 mg por 1 kg de peso do paciente).
A fase profunda da crise hemolítica deve ser interrompida pela transfusão de hemácias após a realização do teste de Coombs. Para aliviar os distúrbios hemodinâmicos que frequentemente acompanham o curso da hemólise aguda, recomenda-se a administração intravenosa de Reogluman na dose calculada de 15 ml por 1 kg de peso do paciente.
A presença de sinais de aumento de uréia e creatinina em um paciente é a base para a hemodiálise. Deve-se levar em consideração que uma violação da técnica e uma alteração na composição do fluido dialisante podem, por si só, provocar o desenvolvimento de uma reação hemolítica intensificada.
Para prevenir o desenvolvimento de insuficiência renal, os pacientes com hemólise devem receber prescrição de bicarbonato de sódio na dose de 5 g com administração oral simultânea de Diacarb na dose de 0,25 g.
O tratamento medicamentoso da hemólise em recém-nascidos consiste na transfusão de reposição primária de sangue Rh negativo. O cálculo da quantidade necessária de sangue administrado é de 150 ml/kg de peso corporal. A transfusão de sangue deve ser combinada com terapia adequada com glicocorticosteróides (administração intramuscular de cortisona na dose de 8 mg curso de curta duração). Os sinais de danos às estruturas do sistema nervoso central são nivelados após o uso de ácido glutâmico na dose de 0,1 g por via oral.
Os métodos não medicamentosos para prevenir a recorrência de hemólise em recém-nascidos incluem evitar a amamentação.
Hemólise - qual médico você deve contatar?? Se você tiver ou suspeitar do desenvolvimento de hemólise, procure imediatamente orientação médica, como hematologista ou transfusiologista.
A anemia hemolítica (AH) é um grupo de doenças heterogêneas unidas por uma única característica patogenética: encurtamento da vida útil dos eritrócitos, desenvolvimento de hemólise dos eritrócitos graus variantes intensidade.
A etiologia e patogênese dessas doenças são diferentes, mas o principal complexo de sintomas clínicos é o mesmo: anemia hiperregenerativa, distúrbios do metabolismo da bilirrubina devido à fração indireta, síndrome hepatolienal. Para estabelecer o diagnóstico, é necessário realizar diagnósticos diferenciais com diversas doenças, inclusive aquelas associadas a distúrbios do metabolismo da bilirrubina.
objetivo comum— ser capaz de diagnosticar AH, navegar pelas formas nosológicas de AH e determinar táticas de manejo do paciente.
Identifique os principais sinais do complexo de sintomas clínicos de hemólise, coloque diagnóstico clínico, determinar as táticas de manejo do paciente, prestar primeiros socorros de emergência para hemólise grave.
Questões teóricas
1. Classificação do GA.
2. Características clínicas do principal complexo de sintomas da HA.
3. AH hereditário: etiologia, patogênese, quadro clínico, táticas de tratamento.
4. AH adquirido: etiologia, patogênese, quadro clínico, táticas de tratamento.
A classificação de anemia mais utilizada até o momento é a proposta em 1979 por L.I. Idelson:
- anemia associada à perda de sangue;
- anemia devido a hematopoiese prejudicada;
- anemia devido ao aumento da destruição sanguínea.
A. Hereditário:
1. Membranopatia (microesferocitose, eliptocitose, piropoiquilocitose, acantocitose, estomatocitose, hemoglobinúria paroxística noturna).
2. Enzimopatias (defeitos do ciclo de Embden-Meyerhoff, ciclo das pentoses fosfato, metabolismo de nucleotídeos, metemoglobinemia).
3. Anemia hemolítica por defeitos na estrutura e síntese da hemoglobina (doença falciforme, talassemia, eritroporfiria).
B. Comprado:
1. Imune e imunopatológica (anemias autoimunes, isoimunes, transimunes, induzidas por haptenos).
3. Anemia hemolítica causada por danos químicos aos glóbulos vermelhos (envenenamento por metais pesados, veneno de cobra).
4. Deficiência de vitaminas (anemia por deficiência de vitamina E da prematuridade).
5. Anemia hemolítica causada por dano mecânico eritrócitos (anemia hemolítica microangiopática, púrpura trombocitopênica trombótica, síndrome hemolítico-urêmica, coagulação intravascular disseminada (DIC), fragmentação de eritrócitos combinada com patologia intracardíaca como resultado de lesão mecânica direta de eritrócitos quando entram em contato com uma prótese valvar, hematomas gigantes (síndrome de Kasabach - Merrita), hemangioma hepático).
Com base na classificação acima, podemos concluir que a GA pode ser tanto uma doença independente quanto um sintoma da doença.
A hemólise dos eritrócitos pode ocorrer de forma aguda, crônica e na forma de crise hemolítica (hemólise aguda) no contexto da hemólise crônica.
As principais características clínicas da hemólise eritrocitária:
- anemia de gravidade variável;
- perturbação do metabolismo da bilirrubina devido ao aumento da fração indireta como resultado da sobrecarga funcional do fígado com produtos de degradação da hemoglobina;
— síndrome hepatolienal com aumento predominante do baço devido ao aumento da carga funcional do fígado e ao aumento da função de sequestro do baço.
Durante uma crise hemolítica, os pacientes queixam-se de fraqueza geral, aumento da temperatura corporal, dor de cabeça, perda de apetite, náuseas, às vezes vômitos, dor abdominal ou sensação de peso no hipocôndrio esquerdo, icterícia, pele pálida com tonalidade cerosa.
Durante um estudo objetivo, o estigma da disembriogênese pode chamar a atenção: crânio em torre, palato gótico, deformação dos maxilares, dentes, hipercromia da íris, retração da ponte nasal, microftalmia, torcicolo, etc.
A anemia é de natureza hiperregenerativa devido à irritação da hematopoiese eritróide para compensar a hemólise. Sinal de laboratório a hemólise dos eritrócitos (aguda ou crônica) é o aumento da reticulocitose, o aparecimento de normócitos devido à liberação da medula óssea de elementos eritróides imaturos contendo núcleos contendo restos de núcleos pertencentes à classe dos elementos eritróides em maturação.
Todas as outras manifestações que podem ocorrer com GA são causadas por uma doença contra a qual ocorre hemólise dos glóbulos vermelhos. Assim, na fase inicial do diagnóstico do AH, é necessário realizar o diagnóstico diferencial com as seguintes doenças:
— AH adquirida e congênita e hemoglobinopatias;
— patologia hepática;
- doenças mieloproliferativas;
- doenças infecciosas.
Anemias hemolíticas hereditárias— grupo grande doenças que combinam HA hereditária associada à ruptura da membrana eritrocitária, fermentopatia eritrocitária e anemia associada à instabilidade da hemoglobina.
Quadro clínico. O AH hereditário, além da síndrome hemolítica de gravidade variável, ocorrendo de forma crônica ou aguda na forma de crise, apresenta características fenotípicas comuns: crânio em torre, ponte nasal recuada, palato gótico, deformação dos maxilares, dentes, prognatismo, sindactilia, polidactilia , pode ser observada microftalmia, íris heterocrômica, torcicolo. Os sinais radiológicos de expansão da cabeça de ponte da medula óssea da hematopoiese são o sintoma de “escova” nas radiografias do crânio e o espessamento da placa interna do osso frontal.
Vamos discutir formas nosológicas individuais de doenças hereditárias que têm maior significado clínico.
Anemia hemolítica microesferocítica hereditária(Doença de Minkowski-Choffard) é uma doença genética (o tipo de herança é autossômica dominante), acompanhada de hemólise de intensidade variável, diminuição da resistência osmótica dos eritrócitos, esferocitose, esplenomegalia e icterícia.
Etiologia. O aumento da destruição de eritrócitos é o resultado de uma deficiência ou patologia de uma ou mais proteínas da membrana eritrocitária (defeito de espectrina e akirina, etc.).
Fisiopatologia:
1. Perda de lipídios na membrana dos glóbulos vermelhos.
2. Desequilíbrio de sódio nos glóbulos vermelhos (aumento do acúmulo de água neles).
3. Redução da área dos glóbulos vermelhos e compactação do citoplasma (reduz a capacidade dos glóbulos vermelhos se deformarem ao passar pelos seios esplênicos).
Os glóbulos vermelhos danificados são engolfados pelos macrófagos esplênicos.
Quadro clínico. O curso da doença é ondulante, a crise hemolítica é substituída por uma remissão relativa que dura de vários meses a 1-2 anos. A crise hemolítica pode ser provocada por infecção, estresse psicoemocional, atividade física, mudando a zona climática. O curso da doença pode ser leve (sem crises ou com frequência de crises não superior a 1 vez em 1-2 anos), moderado (crises 2-3 vezes por ano) e grave com crises frequentes e perturbações graves do metabolismo da bilirrubina .
Características da microesferocitose em crianças pequenas:
- início gradual da doença, progressão lenta da anemia, muitas vezes grave;
- perturbação grave do metabolismo da bilirrubina;
- desenvolvimento frequente de hepatite parenquimatosa;
- em crianças dos primeiros 3 meses, a microesferocitose e a reticulocitose são levemente expressas e aparecem em mais idade avançada. O aparecimento de normoblastos é característico, principalmente durante uma crise;
— a recuperação da crise é lenta;
— juntamente com uma diminuição na resistência osmótica mínima dos eritrócitos, observa-se um aumento na resistência osmótica máxima dos eritrócitos.
Complicações. Em recém-nascidos - kernicterus, em crianças com mais de 1 mês - colelitíase, hepatite crônica, cirrose hepática. Com transfusões de sangue frequentes em pessoas com doença grave - hemossiderose. Crises regenerativas durante a infecção por parvovírus.
Características clínicas das crises regenerativas:
- observado em crianças de 3 a 11 anos, com duração de 4 a 5 dias a 2 semanas;
- início agudo de crise com reação a alta temperatura, intoxicação grave;
— ausência completa de icterícia da pele e esclera;
— o tamanho do baço não aumenta de acordo com a gravidade da anemia;
— no início e no auge da crise hemolítica não há reticulocitose;
- alguns pacientes podem apresentar trombocitopenia;
- no mielograma - inibição da hematopoiese com estreitamento predominante da linhagem eritróide (o processo é reversível).
1. Anemia de gravidade variável. Em 25% dos pacientes, a anemia pode não ser observada devido à compensação. O volume médio de um eritrócito, o conteúdo médio de hemoglobina em um eritrócito e o índice de cor podem ser normais, aumentados ou diminuídos.
2. Reticulocitose grave.
3. O número de leucócitos e plaquetas é normal, aumenta após esplenectomia.
4. Os esfregaços de sangue contêm microesferócitos únicos (eritrócitos menores, hipercromáticos, sem clareamento central, poiquilocitose).
5. Diminuição do volume médio dos eritrócitos durante a eritrocitometria e deslocamento da curva eritrocitométrica para a esquerda.
6. Resistência osmótica reduzida dos eritrócitos: os eritrócitos hemolisam rapidamente em uma solução hipotônica de cloreto de sódio (a hemólise começa em uma solução de 0,6-0,7%).
7. A concentração de bilirrubina no soro sanguíneo aumenta devido à fração indireta de gravidade variável.
8. No mielograma - inibição da hematopoiese com estreitamento predominante da linhagem eritróide (o processo é reversível).
Tratamento. O método de escolha para o tratamento da anemia de Minkowski-Choffard é a esplenectomia. A esplenectomia não é indicada para pacientes com evolução assintomática da doença. Para prevenir complicações infecciosas graves antes ou depois da esplenectomia, recomenda-se a vacinação profilática com vacina antipneumocócica.
Em caso de crise hemolítica, transfusão de sangue de reposição conforme indicações vitais na dose de 8-10 mg/kg, terapia de desintoxicação, correção do equilíbrio hídrico e eletrolítico, medicamentos cardiovasculares conforme indicações.
Glicocorticóides e suplementos de ferro não são aconselháveis. Durante uma crise regenerativa, os corticosteróides de curta duração são indicados na dose de 1-1,5 mg/kg.
Anemia hemolítica hereditária não esferocítica associada à deficiência de enzimas eritrocitárias,- um grupo heterogêneo de anemias resultantes da perturbação de vários sistemas enzimáticos de utilização da glicose, a maioria dos quais são acompanhados por hemólise crônica ou intermitente com alterações inespecíficas na morfologia dos eritrócitos: basofilia, policromasia, esferocitose, eritrócitos semelhantes a alvo. Este grupo de doenças é caracterizado por:
— resistência osmótica normal no sangue incubado;
- aumento da auto-hemólise de sangue estéril incubado a uma temperatura de 37 ° C (normalmente, após 48 horas, a porcentagem de lise eritrocitária é de 0,4-4,5%; com esse tipo de anemia hemolítica, até 40% dos eritrócitos podem ser hemolisados);
- metabolismo defeituoso dos eritrócitos.
Quadro clínico AH hereditário não esferocítico: episódios de hemólise após exposição a oxidantes ou infecção; HA crônica; hemólise aguda após ingestão de feijão (favismo); metemoglobinopatia; icterícia de recém-nascidos.
A anomalia mais comum dos glóbulos vermelhos é deficiência de atividade da glicose-6-fosfato desidrogenase (G-6-PDH). O gene estrutural responsável pela síntese de G-6-FDG está localizado no cromossomo X; o locus está localizado próximo ao gene do daltonismo e, portanto, é frequentemente combinado com o daltonismo. O tipo de herança é incompletamente dominante, ligado ao sexo. Segundo isso, os meninos são homozigotos e sofrem desta doença. As meninas homozigotas adoecem, as heterozigotas têm 50% de atividade enzimática e não adoecem.
Existem formas africanas, mediterrânicas e raras de deficiência grave de G-6-FDG.
Patogênese. As células deficientes em G-6-FDG são limitadas na sua capacidade de gerar NADP e formar uma forma reduzida de glutationa, que é necessária para reduzir o conteúdo de peróxido de hidrogénio e radicais livres que surgem durante a função celular. A explosão de oxigênio resultante do excesso de peróxido de hidrogênio leva à desnaturação da proteína que está ligada à membrana dos glóbulos vermelhos. Os chamados corpos de Heinz resultantes alteram a forma e a estrutura dos glóbulos vermelhos. Quando os glóbulos vermelhos passam pelo fígado e pelo baço, os corpúsculos de Heinz, juntamente com parte membrana celular“beliscados” por macrófagos.
Quadro clínico Deficiência de G-6-FDG. Em recém-nascidos, a HA é frequentemente grave e requer transfusão de sangue de reposição. Com a maturação do sistema glicuroniltransferase hepático, o grau de hiperbilirrubinemia diminui.
Em crianças maiores e adultos, a deficiência de G6-FDG manifesta-se como HA crônica, que geralmente é agravada pela adição de doenças e/ou medicamentos intercorrentes.
Medicamentos que causam hemólise dos glóbulos vermelhos devido à deficiência de G-6-FDG: medicamentos antimaláricos, sulfonamidas, nitrofuranos, analgésicos, produtos químicos - azul de metileno, naftaleno, fenilhidrazina, trinitrotolueno, etc. A hemólise aguda ocorre no segundo dia de uso dos medicamentos. O quadro clínico é representado por hemólise aguda, insuficiência renal aguda e, em alguns pacientes, síndrome DIC. A retirada dos medicamentos leva à cessação da hemólise. No hemograma, além dos sinais característicos da hemólise, observam-se neutrofilia com desvio para a esquerda e granularidade tóxica dos neutrófilos.
A correção da acidose durante a infecção interrompe a hemólise.
Uma das manifestações mais graves da deficiência de G-6-FDG é o favismo. Ocorre em crianças de 1 a 5 anos quando comem favas ou inalam favas. A hemólise aguda aparece 5 a 24 horas após a ingestão do feijão. Há palidez acentuada da pele e das membranas mucosas, febre, hemoglobinúria, dor nas costas, hemoglobina (Hb) diminui para 60-40 g/l. Muitas vezes complicado por insuficiência renal aguda. Após 3-4 dias do início da hemólise, ocorre uma recuperação lenta.
Diagnóstico laboratorial. Durante uma crise: anemia grave, leucocitose com desvio para a esquerda. Morfologia dos eritrócitos durante a crise: presença de corpos de Heinz, células fragmentadas. Após 4-5 dias, a reticulocitose aparece com pico após 10-20 dias.
Anemia macro ou microcítica; esfregaços de sangue periférico mostram cor e formato anormais e presença de corpos de Heinz. Com hemólise intravascular maciça - hemoglobinúria. O diagnóstico de deficiência de G-6-FDG deve basear-se em definição direta atividade enzimática. É indicada a determinação da atividade G-6-FDG em familiares do paciente.
Tratamento. Suspensão da medicação que causou a crise. Tratamento da infecção, descompensação da diabetes mellitus, no contexto da qual surgiu uma crise. Em recém-nascidos com hiperbilirrubinemia grave, é realizada uma transfusão de sangue de reposição. Terapia de desintoxicação, correção do volume sanguíneo circulante, equilíbrio ácido-base.
As transfusões de sangue de reposição são utilizadas apenas em casos de anemia grave no contexto de anticoagulantes (a hemólise maciça dos glóbulos vermelhos leva à liberação de substâncias tromboplásticas e provoca a síndrome da coagulação intravascular disseminada). Em caso de hemólise intravascular maciça, está indicada a plasmaférese, em caso de insuficiência renal aguda - hemodiálise.
As vacinações preventivas são realizadas apenas por indicações epidemiológicas.
Talassemia- um grupo de doenças com distúrbio hereditário na síntese de uma ou mais cadeias de globina. Devido a um desequilíbrio na produção de cadeias de globina, desenvolvem-se hematopoiese ineficaz, produção defeituosa de Hb, hemólise e anemia de gravidade variável.
Epidemiologia. As hemoglobinopatias são as doenças monogênicas mais comuns doenças hereditárias em crianças (cerca de 240 milhões de pessoas na Terra, segundo a Organização Mundial da Saúde). Todos os anos, cerca de 200 mil pessoas nascem e morrem no mundo com esta doença. As hemoglobinopatias são frequentemente detectadas na Transcaucásia, Ásia Central, Daguestão, Moldávia, Bashkiria, etc.
Fisiopatologia. Cada molécula de Hb consiste em 2 pares separados de cadeias de globina idênticas. Nos adultos, a Hb é representada pela HbA (96%) e HbA2 (2,5%). No feto, predomina a HbF fetal. Vários tipos de talassemias estão associados a um defeito em qualquer uma das cadeias polipeptídicas da globina. A deficiência seletiva de uma ou mais cadeias polipeptídicas de globina tem duas consequências imediatas:
- diminuição da síntese de Hb;
— desequilíbrio na síntese das cadeias de globina com aparecimento de quantidade excessiva de cadeias de globina;
- perturbação dos processos metabólicos no eritrócito. Este último revela-se funcionalmente inferior e é destruído nas células do sistema reticuloendotelial, desenvolvendo-se eritropoiese ineficaz e utilização prejudicada do ferro formado como resultado da quebra da Hb.
Dependendo do grau de redução na síntese de uma ou outra cadeia polipeptídica da molécula de Hb, existem 2 tipos principais de talassemia: a e b. Na a-talassemia, a HbA é substituída completamente (na forma homozigótica) ou parcialmente (na forma heterozigótica) por HbF e HbA2. Na b-talassemia, a produção de cadeias b é reduzida ou interrompida. Como a síntese das cadeias a não é prejudicada, neste caso a formação de HbF e HbA2 será mais intensa.
Se o paciente for heterozigoto e um dos alelos mantiver a capacidade de produzir a cadeia B, a quantidade de HbA no sangue será reduzida com o aumento das quantidades de HbF e HbA2 (talassemia menor). Se o paciente for homozigoto, o sangue contém 80-90% de HbF e uma quantidade aumentada de HbA2 (talassemia maior - doença de Cooley).
Quadro clínico depende da homo ou heterozigosidade. De acordo com a gravidade, distinguem-se a talassemia maior, a talassemia menor e a talassemia mínima. A talassemia maior (doença de Cooley) é mais comum em homozigotos com talassemia b e é caracterizada por uma diminuição acentuada de até 10% na HbA com um aumento significativo na HbF fetal. Caracteriza-se por anemia progressiva com eritroblastemia, hepatoesplenomegalia, aumento da hemólise com urobilirrubinemia, mas sem pigmentos biliares na urina, osteoporose com formação de esqueleto facial mongolóide, sintoma de “escova”, crânio em torre, palato gótico (expansão das cabeças de ponte da hematopoiese). Há um atraso no desenvolvimento mental e mental, febre, icterícia leve e coloração acinzentada da pele devido à hemossiderose, são observadas periodicamente pigmentação marrom da pele. O abdômen aumenta acentuadamente de tamanho devido ao tamanho colossal do fígado e do baço. De acordo com o curso, existem formas fulminantes, crônicas e crônicas prolongadas, nas quais o paciente sobrevive até a puberdade.
A talassemia menor ocorre em portadores heterozigotos da característica. É caracterizada pelos mesmos sintomas do grande, mas menos pronunciados. A doença é grave em tenra idade. Infecções intercorrentes e estresse podem levar a uma crise hemolítica. Às vezes, os únicos sinais de doença podem ser alterações laboratoriais.
Diagnóstico laboratorial. Principal critério diagnósticoé a identificação de HbF, A2, H no estudo dos tipos de hemoglobina. Na análise do sangue periférico na talassemia maior, anemia microcítica hipocrômica grave, eritroblastose, normoblastose, reticulocitose. O esfregaço revela glóbulos vermelhos em formato de alvo. A resistência osmótica dos eritrócitos é alta (a hemólise pode ocorrer mesmo em solução de cloreto de sódio 0,1-0,2%).
No contexto de transfusões de sangue frequentes, o nível de ferro e ferritina séricos aumenta. Na radiografia: osteoporose, sintoma de “escova”, vértebras de “peixe”.
Tratamento. Para talassemia maior - transfusões de sangue de reposição freqüentes de 15 ml/kg uma vez a cada 4-5 dias. Complicações: hemossiderose, que requer terapia com deferoxamina (Desferal), exijade para reduzir a sobrecarga de ferro. A esplenectomia é ineficaz. Um tratamento radical para a talassemia maior é o alotransplante de medula óssea.
Anemia falciforme. O termo “doença falciforme” é utilizado para designar um processo patológico em que se observa anemia devido ao transporte de Hb, que altera sua estrutura em condições de hipóxia.
Etiologia e patogênese. A anemia falciforme é uma doença na qual a HbS anormal é sintetizada. Na cadeia B, a molécula de ácido glutâmico é substituída por uma molécula de valina, o que leva a uma alteração nas propriedades da molécula da proteína globina. Esta pequena mudança na estrutura é responsável por profundos distúrbios na estabilidade e solubilidade molecular. A carga elétrica da Hb muda, os glóbulos vermelhos perdem a capacidade de se desconfigurar, aderir e hemolizar sob condições hipóxicas. Uma diminuição acentuada na solubilidade da HbS sob condições hipóxicas leva à deformação crescente dos eritrócitos, aumento da viscosidade do sangue, estase vascular, adesão dos eritrócitos ao endotélio, danos nos tecidos e isquemia de órgãos, que se manifesta clinicamente por dor.
A capacidade dos glóbulos vermelhos de formar foices é proporcional ao conteúdo de HbS. Pacientes cujos glóbulos vermelhos contêm menos de 50% de HbS não apresentam sintomas da doença. A falcização aumenta com a acidose e diminui com a alcalose. Nos seios da face do baço, os glóbulos vermelhos falciformes são hemolisados.
As crises podem ser desencadeadas por infecção, desidratação durante a febre, acidose durante o jejum e hipóxia durante diversas doenças.
Herança e epidemiologia. O gene falciforme é comum nos países do Oriente Médio, Grécia, Índia, mas mais frequentemente na África tropical com uma frequência de heterozigosidade superior a 40%. Foi observada uma combinação geográfica da doença falciforme com áreas onde a malária é endémica. Nos homozigotos com HbSS observa-se o quadro mais clássico da doença falciforme, na forma heterozigótica fala-se em anomalia falciforme.
Quadro clínico. Nos recém-nascidos, níveis elevados de HbF desempenham um papel protetor durante as primeiras 8 a 10 semanas. Quando uma criança tem 3 meses ou mais, as crises são mais frequentemente desencadeadas pelo ARVI, várias condições acompanhada de hipóxia, anestesia, etc.
Existem vários tipos de crises na doença falciforme.
Crise vaso-oclusiva: pode ocorrer no contexto de fatores provocadores diariamente e várias vezes por ano. Caracterizada por hipóxia tecidual e infartos de órgãos devido à microcirculação prejudicada por eritrócitos em forma de foice. Acompanhada pelo desenvolvimento de paralisia (estase nos vasos cerebrais), hematúria (estase nos capilares renais), necrose óssea asséptica, úlceras cutâneas, cardiomegalia, mialgia, infarto pulmonar, fígado, baço. Crises frequentes podem causar fibrose esplênica, asplenia funcional e cirrose hepática. Todos os casos são caracterizados por dor intensa. A crise geralmente se resolve dentro de algumas horas a alguns dias.
Crise de sequestro: aparece em crianças e muito raramente em adultos. Devido a causas desconhecidas, pacientes com esplenomegalia significativa apresentam sequestro passivo repentino de glóbulos vermelhos no baço, o que pode causar hipotensão e levar à morte súbita.
Crise hemolítica: Sempre é observada hemólise moderada constante dos glóbulos vermelhos, mas às vezes pode ocorrer hemólise maciça súbita com uma diminuição acentuada da Hb (raramente observada).
Crise aplástica: mais frequentemente causada por parvovírus humano B19 e condições acompanhadas de deficiência de folato.
As crianças com HbSS homozigótica tendem a ser baixas e a atrasar a puberdade, mas o seu crescimento continua até ao final da adolescência e atinge níveis normais na idade adulta. Todos os estigmas de HA hereditário são característicos devido à expansão da cabeça de ponte da hematopoiese (face mongolóide, crânio em torre, sintoma de “escova” nas radiografias do crânio, vértebras “peixe”). Muitas vezes ocorre após 4 anos necrose asséptica cabeças femorais. Fraturas ósseas espontâneas são possíveis. O priapismo é detectado em meninos. As complicações da doença são danos ao fígado (colestase, cirrose, colelitíase), rins (hipostenúria, hematúria), fibrose esplênica e asplenia funcional, várias lesões do sistema nervoso central.
Diagnóstico laboratorial. O principal método de diagnóstico é a eletroforese de hemoglobina, que revela aumento dos níveis de HbS. Quando combinado anemia falciforme e b-talassemia, aumento das concentrações de HbF e HbA2. No exame de sangue: anemia normocítica normocrômica de gravidade variável, anisocitose, poiquilocitose; em crise, podem ser detectados eritrócitos falciformes, eritrócitos alvo, reticulocitose moderada. Leucocitose e trombocitose são frequentemente observadas devido à demarcação do pool periférico de leucócitos no contexto de distúrbios microcirculatórios, aumento da função da medula óssea e asplenia funcional.
Tratamento. Tratamento eficaz Não existe doença falciforme, portanto o cuidado ao paciente deve ser direcionado ao tratamento de complicações.
Para aumentar a eritropoiese, os suplementos de ácido fólico devem ser prescritos por um longo período. As transfusões de glóbulos vermelhos não são realizadas rotineiramente, mas as transfusões sanguíneas profiláticas podem reduzir o número de crises, mas o risco da transfusão em si pode ser maior.
Durante uma crise, o paciente deve ser mantido aquecido e receber hidratação adequada e analgésicos, e o oxigênio é eficaz. A anestesia geral deve ser utilizada com muito cuidado devido ao alto risco de crise hemolítica.
O HA isoimune é caracterizado pelo fato de a hemólise dos eritrócitos ocorrer sob a influência de anticorpos contra os antígenos eritrocitários do paciente que entram no corpo vindos de fora (doença hemolítica do recém-nascido; HA autoimune na mãe; transfusão de eritrócitos incompatíveis com o sistema ABO , fator Rh, etc.). O HA heteroimune está associado ao aparecimento de um novo antígeno na superfície do eritrócito do paciente. Esse novo antígeno pode ser um medicamento que o paciente recebe (antibiótico, sulfonamida, etc.), antígenos de vacinas preventivas. O complexo antígeno-anticorpo é fixado na membrana eritrocitária, o hapteno também pode ser um vírus (vírus Epstein-Barr, etc.). A hemólise das hemácias ocorre devido à adição de complemento e sua destruição pelos macrófagos. O curso do HA heteroimune geralmente é agudo e termina após a descontinuação da medicação e eliminação da infecção.
Autoimunes são chamados de GAs que ocorrem quando a tolerância imunológica aos antígenos dos eritrócitos do sangue periférico, eritrocariócitos e outros precursores da eritropoiese é perturbada. Todos os AG autoimunes podem ser divididos em idiopáticos e sintomáticos devido à doença geral(colite ulcerativa inespecífica, hepatite autoimune, giardíase.). O AH autoimune é observado em qualquer idade da infância, excluindo os primeiros meses de vida. Assim, a etiologia da doença é variada.
De acordo com a patogênese, distinguem-se HA autoimune com antígenos quentes incompletos, hapteno imune, HA com antígenos frios e HA autoimune com hemolisinas bifásicas em crianças pequenas.
Diagnóstico laboratorial. Determinação do anticorpo anti-eritrocitário por via direta e amostra indireta Coombs. EM análise clínica sangue: anemia moderada/grave, normocrômica, normocítica, reticulocitose. No caso do início mais agudo e agudo - leucocitose, neutrofilia com desvio para a esquerda. Violação do metabolismo da bilirrubina devido à fração indireta.
Tratamento. EM prática pediátricaÉ necessário tratar GA autoimune predominantemente idiopática do tipo térmico. O principal método de tratamento é a monoterapia com esteróides - prednisolona na dose diária de 2 mg/kg, dividida em 2-3 doses. O curso deve ser de pelo menos 4 semanas com retirada gradual sob controle de reticulocitose e com teste de Coombs negativo. Nos casos de GA autoimune resistente aos glicocorticóides, são prescritos imunossupressores: azatioprina (imuran 2-4 mg/kg); ciclofosfamida 2-3 mg/kg com seleção individual de regime e dose. As transfusões de sangue são realizadas apenas por motivos de saúde: hemácias lavadas de acordo com seleção individual.
1.Alekseev N.A. Hematologia e imunologia da infância / N.A. Alekseev. - M.: Hipócrates, 2009. - 1039 p.
2. Guseva S.A. Doenças do sistema sanguíneo / S.A. Guseva, V.P. Voznyuk. - M.: Medpress-Inform, 2004. - 488 p.
3. Guseva S.A. Anemia / S.A. Guseva, Ya.P. Goncharov. - K.: Logos, 2004. - 408 p.
4. Kuzmina L.A. Hematologia da infância / L.A. Kuzmina. - M.: Medpress-Inform, 2001. - 400 p.
5. Guia prático sobre doenças infantis/Ed. A.G. Rumyantseva, E.V. Samochatova. - M.: Medpraktika, 2004. - 792 p.
6. Guia de hematologia laboratorial / Ed. Ed. IA Vorobyova. - M.: Medicina prática, 2011. - 352 p.
7. Sheffman FJ Fisiopatologia do sangue / F.J. Sheffman. - M.: Binom, 2009. - 448 p.
13.09.2017A hemólise não é uma doença, mas um processo que ocorre em um organismo vivo por razões fisiológicas e patogenéticas. A primeira opção é considerada normal e a segunda requer diagnóstico e tratamento precisos. Em palavras simples, hemólise é a morte dos glóbulos vermelhos - um processo destrutivo que ocorre continuamente nos organismos vivos.
O ciclo de vida dos glóbulos vermelhos dura aproximadamente 120 dias, depois a membrana se rompe e a hemoglobina se rompe. Esta é a hemólise fisiológica, pela qual o sistema imunológico é responsável. Essa hemólise não representa perigo para a saúde, ocorre dentro dos limites da normalidade e não causa desconforto ou outros sintomas.
Quando a destruição de glóbulos vermelhos é detectada em sistema circulatório, o processo é denominado hemólise intravascular. A proteína absorve a hemoglobina e segue para o fígado, convertendo a proteína em bilirrubina, que é excretada na bile. Você pode descobrir o que é hemólise sanguínea:
Os motivos listados são classificados como adquiridos, mas também existem fatores congênitos pelos quais os glóbulos vermelhos são destruídos pelos anticorpos do corpo. Esta condição é chamada de hemólise imunológica. Nesta situação, o ciclo de vida de um glóbulo vermelho dura apenas 10 dias. O corpo não tem tempo para repor o número de glóbulos vermelhos.
Outra razão é tomar diuréticos, comprimidos para diabetes, aspirina, malária e tuberculose - esses medicamentos podem destruir os glóbulos vermelhos. O médico que prescreve esses medicamentos deve avisar o paciente quanto tempo dura o curso e como tomar os comprimidos. A automedicação é inaceitável porque não permite o controle do hemograma.
A destruição mecânica dos glóbulos vermelhos ocorre no contexto de um trauma significativo no corpo humano, da presença de um marcapasso e também de choque elétrico.
A natureza não é apenas amiga do homem, mas ao mesmo tempo uma fonte de perigo se você não souber se comportar corretamente. Por exemplo, a hemólise dos glóbulos vermelhos às vezes é provocada pelos seguintes fatores:
Os motivos listados acima causam hemólise não instantaneamente, mas à medida que o fator provocador atua. Em qualquer um desses casos é necessária consulta médica e tratamento visando prevenir complicações.
Se a pessoa não apresentar sintomas de hemólise atípica e a destruição das hemácias não estiver associada à patologia e ocorrer conforme planejado, a pessoa não sentirá nenhum sintoma, e isso é normal, pois a hemólise natural é proporcionada pela natureza.
As manifestações de hemólise são detectadas apenas em casos de natureza e origem atípica da patologia. A patologia ocorre de forma aguda e subcompensada, após o qual se inicia um período de remissão.
A hemólise aguda dos eritrócitos é caracterizada por um rápido desenvolvimento e o estado de saúde de uma pessoa deteriora-se acentuadamente. Na maioria das vezes, ocorre um quadro agudo após uma transfusão de sangue, se os componentes não couberem, bem como em caso de intoxicação ou infecção por medicamentos. A insidiosidade da hemólise aguda reside na intensidade das manifestações, e o corpo simplesmente não consegue restaurar o número de glóbulos vermelhos em vez dos destruídos. Portanto, a hemólise dos eritrócitos na forma aguda se manifesta por anemia e intoxicação por bilirrubina.

No curso agudo da doença, a pele fica amarelada e o paciente começa a sentir náuseas e vômitos.
Na hemólise aguda, a pele fica amarelada, o paciente queixa-se de náuseas e vômitos. A dor é sentida no abdômen, mas a pessoa não consegue indicar com precisão sua localização. A patologia grave é acompanhada por desmaios e convulsões. A anemia é acompanhada por fraqueza, palidez e falta de ar. O baço aumenta e o mesmo acontece com o fígado. A urina fica escura. Os resultados dos testes revelam alterações na composição do sangue e da urina - são determinadas hemoglobinemia e bilirrubinemia graves, trombocitopenia. Ao mesmo tempo, o nível de uréia e creatinina aumenta, os fatores de fibrinólise e hemoglobinúria diminuem.
A forma aguda não é perigosa por si só, mas por causa de suas complicações. Isto pode ser cardiovascular e insuficiência renal na forma aguda, síndrome DIC.
Quanto à forma subcompensada de hemólise, nesta fase é potencializado o processo de produção de hemácias por um processo especial da medula óssea. Considerando que o número de hemácias é gradativamente compensado, neste caso as manifestações clínicas não são tão marcantes, mas ainda assim bastante distinguíveis. Trata-se de aumento dos parâmetros do fígado e baço, manifestações dermatológicas. Nessa forma, a anemia praticamente não é detectada e os exames laboratoriais mostram aumento no número de reticulócitos, o que indica que está ocorrendo um processo regenerativo no sangue.
Menção separada deve ser feita à hemólise em recém-nascidos. As primeiras manifestações de hemólise começam nas crianças ainda no momento do seu desenvolvimento intrauterino. A causa da hemólise é a incompatibilidade dos parâmetros sanguíneos da mãe e do filho.
A extensão em que a hemólise se manifestará depende diretamente da taxa de aumento nos títulos de anticorpos em sangue materno. Clinicamente, a hemólise em bebês pode se desenvolver de três maneiras:
Para determinar quão alta é a densidade da membrana eritrocitária, é utilizada a técnica de resistência osmótica. O teste permite identificar 2 tipos de destruição de projéteis - mínima e máxima. Considerando o quão prejudicial a solução de NaCl é às hemácias, é no recipiente que a contém que são colocadas as amostras de sangue selecionadas para pesquisa. Quando a concentração da solução resultante varia entre 0,46-0,48%, significa que a densidade das membranas das células sanguíneas é bastante boa e elas não morrem nesta substância. Este teste é chamado de resistência mínima. E a resistência máxima é calculada quando a concentração da solução é de 0,34%. Sob tais condições, todos os glóbulos vermelhos morrem.

Tipos de hemólise
Jovem células sanguíneas são mais resistentes à destruição devido ao seu formato único, mas os glóbulos vermelhos maduros na forma de bolas são rapidamente destruídos. Uma solução hipertônica para determinar a densidade das membranas celulares pode ter diferentes concentrações e, em cada caso, o efeito na atividade vital dos glóbulos vermelhos mudará.
Por exemplo, utiliza-se ativamente solução salina, que não agride o sangue, mas estimula a reposição do volume sanguíneo intravascular. Essa necessidade surge quando sangramento intenso. As membranas dos glóbulos vermelhos da solução de NaCl são fortalecidas e a hemoglobina não se rompe.
A escolha do método de tratamento depende do grau de hemólise, da duração da patologia e do histórico médico do paciente. Em caso de crise hemolítica, medidas urgentes devem ser tomadas na unidade de terapia intensiva ou no setor de internação de hematologia. Será necessária a realização de terapia intensiva e uma série de medidas médicas que possam combater a patologia atual e prevenir tal quadro. Independentemente do que causa a hemólise aguda, o tratamento será mais ou menos assim:

A automedicação não vale a pena, pois primeiro é preciso fazer exames de sangue e, a partir deles, traçar um método de tratamento
Já os recém-nascidos que apresentam conflito Rh com a mãe recebem imediatamente prescrição de transfusão de sangue e terapia hormonal.
Cuidado e atenção básicos ajudarão a prevenir a hemólise causada por fatores externos. Por exemplo, você não pode colher e comer frutas e cogumelos desconhecidos, e o mesmo se aplica a plantas desconhecidas. Se você for picado por uma aranha ou outro inseto, será necessário cauterizar a área afetada e espremer o veneno. Isso minimiza a quantidade de toxinas no sangue e melhora o bem-estar da pessoa. A automedicação não é estritamente recomendada, pois é necessário estudar exames de sangue para selecionar o método e os medicamentos.

O sentido da vida está relacionado com a questão “Para que viver”, e não com a questão de como manter a vida. A atitude de uma pessoa é...

Um cogumelo é um organismo vivo que forma um reino separado com o mesmo nome. Por muito tempo foram classificados como parte do reino vegetal. Mas em...

Para os amantes da caça “tranquila”, a temporada dos cogumelos começa no início do verão e vai até o final do outono. E raramente o fazem...

Aleshnikova, V.I. Uso de consultores profissionais. - M.: Infra-M, 1999. - 240 p. 2. Beich, E....

Suco de laranja. O significado simbólico do suco de laranja nos livros de sonhos é prazer e tentação. Muitas vezes nós...

Na maioria das vezes superamos todos os tipos de dificuldades encontradas ao longo do caminho da vida. Claro, para isso fazemos...

Este ano o seu patrono Netuno estará na sua constelação e isso é um bom sinal, pois você...

1993 quem? 1993 é o ano de qual animal? — De acordo com o horóscopo chinês, 1873, 1933, 1993 pertenciam aos anos do Negro...

A dualidade onda-partícula da luz significa que a luz tem simultaneamente as propriedades de continuidade...

O papel da biologia é enorme em nosso mundo. Embora não seja uma das disciplinas prioritárias, a maioria dos escolares e...
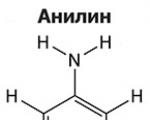
As aminas são derivados orgânicos da amônia contendo um grupo amino NH 2 e um radical orgânico. Em geral...
Como responder às perguntas da Parte BA segunda parte do trabalho de estudos sociais consiste em 7 tarefas com uma resposta curta....

O formulário TORG-15 é elaborado no caso de durante o transporte, movimentação entre e dentro do armazém, quando...

Os nutricionistas dizem que para ter uma boa saúde e um corpo esguio é preciso incluir lanches na sua...

Um cogumelo é um organismo vivo que forma um reino separado com o mesmo nome. Durante muito tempo foram classificados como um reino...

Para os amantes da caça “tranquila”, a temporada dos cogumelos começa no início do verão e vai até o final do outono. E raramente o fazem...