Arenque sob um casaco de pele - uma receita clássica
O que seria do Ano Novo sem champanhe, tangerinas, Olivier, formol e o “arenque com casaco de pele” preferido de todos. Com o último...
Lado esclerose amiotrófica(ELA, doença do neurônio motor, doença de Charcot) é uma patologia bastante rara do sistema nervoso na qual uma pessoa desenvolve fraqueza e atrofia muscular, que inevitavelmente progride e leva a resultado fatal. Você já aprendeu sobre as causas e o mecanismo de desenvolvimento da doença com o anterior, vamos agora falar sobre os sintomas, métodos de diagnóstico e tratamento da ELA.
 A fraqueza muscular progressiva nas pernas leva à incapacidade do paciente de se mover de forma independente.
A fraqueza muscular progressiva nas pernas leva à incapacidade do paciente de se mover de forma independente.
Para descobrir qual manifestações clínicas pode ter a doença de Charcot, você deve entender o que são neurônios motores centrais e periféricos.
O neurônio motor central está localizado no córtex hemisférios cerebrais. Se for afetado, desenvolve-se fraqueza muscular (paresia) em combinação com aumento tônus muscular, intensificam-se os reflexos, que são verificados com um martelo neurológico durante o exame, aparecem sintomas patológicos (reação específica dos membros a certas irritações, por exemplo, extensão do 1º dedo do pé ao irritar a borda externa do pé, etc.).
O neurônio motor periférico está localizado no tronco cerebral e em vários níveis medula espinhal(cervical, torácica, lombossacra), ou seja, abaixo do central. Com a degeneração desse neurônio motor, também se desenvolve fraqueza muscular, mas é acompanhada por diminuição dos reflexos, diminuição do tônus muscular, ausência de sintomas patológicos e desenvolvimento de atrofia dos músculos inervados por esse neurônio motor.
O neurônio motor central transmite impulsos ao periférico, que transmite impulsos ao músculo, e o músculo se contrai em resposta a isso. No caso da ELA, em algum momento a transmissão do impulso é bloqueada.
Na esclerose lateral amiotrófica, tanto os neurónios motores centrais como os periféricos podem ser afectados, em várias combinações e em diferentes níveis (por exemplo, há degeneração do neurónio motor central e do neurónio motor periférico ao nível cervical ou apenas do neurónio motor periférico ao nível lombossacro nível no início da doença). É isso que determina quais sintomas o paciente terá.
Destaque seguintes formulários BAIXO:
Esta classificação baseia-se na determinação dos sinais predominantes de lesão de qualquer um dos neurônios no início da doença. À medida que a doença continua a existir, perde o seu significado porque processo patológico cada vez mais neurônios motores estão envolvidos em diferentes níveis. Mas tal divisão desempenha um papel no estabelecimento de um diagnóstico (será que é ELA?) e na determinação do prognóstico para a vida (quanto tempo se espera que o paciente viva).
Os sintomas comuns característicos de qualquer forma de esclerose lateral amiotrófica são:
Com esta forma da doença, duas opções são possíveis:

Também pode estrear de duas maneiras:
A esclerose lateral amiotrófica (ELA) é uma doença incurável e progressiva do sistema nervoso central, na qual o paciente sofre danos nos neurônios motores superiores e inferiores, o que causa atrofia muscular e paralisia. A frequência desta patologia é de cerca de 2 a 7 casos por 100 mil pessoas. Na maioria das vezes, a doença é diagnosticada em pacientes com mais de 50 anos de idade.
Os cientistas ainda não criaram uma classificação unificada e abrangente de ELA. Existem várias abordagens para classificar a doença. Por exemplo, a abordagem norte-americana envolve a identificação dos seguintes tipos de ELA: esporádica, familiar, endêmica esporádica. A classificação da esclerose lateral amiotrófica prevê as seguintes formas da doença: bulbar, lombossacra, cervicotorácica e primária generalizada. Existem também diversas variantes da doença: mista, piramidal e segmentar-nuclear.
Para o mais comum sintomas iniciais As doenças incluem cãibras (espasmos musculares dolorosos), letargia e fraqueza nos braços distais, distúrbios bulbares, atrofia muscular das pernas, fraqueza nos cintura escapular. Além disso, diferentes variantes da doença são caracterizadas por diferentes manifestações clínicas.
As causas exatas da esclerose lateral amiotrófica ainda estão sendo investigadas pelos cientistas. Porém, podem ser citados vários fatores que provocam a doença. Por exemplo, cerca de 5% das doenças têm etiologia hereditária. Pelo menos 20% dos casos estão associados a mutações no gene da superóxido dismutase-1. Os cientistas provaram que papel importante A alta atividade do sistema glutamatérgico desempenha um papel no aparecimento da doença. O fato é que o excesso de ácido glutâmico provoca superexcitação e morte súbita dos neurônios. O mecanismo genético molecular da patologia também foi comprovado. É causada por um aumento no nível de DNA e RNA nas células, o que acaba levando à interrupção da síntese protéica.
Os cientistas também identificam vários fatores predisponentes que desempenham um papel importante na ocorrência de ELA. Em primeiro lugar, esses fatores incluem a idade. O fato é que a doença geralmente se desenvolve em pacientes com idade entre 30 e 50 anos. Vale lembrar que apenas cerca de 5% dos pacientes apresentam predisposição hereditária para o BAS. Na grande maioria dos casos de ELA, a causa da patologia não pode ser determinada.
O curso inicial da doença é caracterizado por sintomas como convulsões, espasmos, dormência muscular, dificuldade para falar e fraqueza nos membros. Como esses sintomas são típicos de muitos doenças neurológicas, diagnosticar ELA em estágio inicial difícil. Na maioria dos casos, a doença pode ser diagnosticada na fase de atrofia muscular.
Dependendo da doença afetada partes diferentes corpo, distinguir ELA de membros e ELA bulbar. No primeiro caso, os pacientes apresentam deterioração da flexibilidade do tornozelo, falta de jeito ao caminhar e começam a tropeçar. A ELA bulbar se manifesta por dificuldade para falar (som nasal, dificuldade para engolir). Logo o paciente sente dificuldade para se movimentar ou não consegue mais se movimentar de forma independente. Normalmente, a doença não afeta negativamente as habilidades mentais do paciente, mas leva à depressão grave. Na maioria dos casos, decorrem cerca de três a cinco anos desde o aparecimento dos primeiros sintomas até à morte.
Como a ELA é uma doença incurável que encurta rapidamente a vida de uma pessoa, o exame do paciente deve ser abrangente e preciso. É extremamente importante colocar diagnóstico correto o paciente para iniciar o alívio oportuno de seus principais sintomas, pois isso pode prolongar a vida do paciente. O plano de exame geralmente inclui história de vida e doença, exame neurológico e físico, ressonância magnética da medula espinhal e do cérebro, EMG e exames laboratoriais.
O diagnóstico da doença começa com uma entrevista detalhada com o paciente. Ou seja, o médico precisa esclarecer se o paciente se queixa de espasmos e espasmos musculares, fraqueza e rigidez, dificuldade de movimento das mãos, fala, caminhada, deglutição, salivação, falta de ar frequente, perda de peso, fadiga, falta de ar durante a execução exercício físico. Além disso, o médico deve perguntar se o paciente notou visão dupla, perda de memória, sensação de formigamento pelo corpo ou problemas urinários. É imprescindível perguntar ao paciente sobre sua história familiar - se ele tem parentes com distúrbios crônicos do movimento.
O principal objetivo do exame físico é avaliar a constituição do paciente, pesá-lo, medir sua altura e calcular seu índice de massa corporal. O exame neurológico geralmente inclui testes neuropsicológicos. Ao avaliar as funções bulbares, o médico atenta para o timbre da voz, velocidade da fala, reflexo faríngeo, presença de atrofias da língua e paresia do palato mole. Além disso, durante o exame, é verificada a força dos músculos trapézios.
O principal método instrumental para diagnosticar a doença é a EMG de agulha. Essa técnica permite identificar sinais da doença, como desnervação aguda ou crônica. Nos estágios iniciais da doença, a estimulação EMG é ineficaz porque não detecta sinais perceptíveis de ELA.
No processo de diagnóstico da doença, os médicos também utilizam métodos de neuroimagem. Grande importância A ressonância magnética da medula espinhal e do cérebro desempenha um papel no diagnóstico diferencial da ELA. Durante a ressonância magnética, em 17-67% dos pacientes é possível identificar sintomas de degeneração dos tratos piramidais e atrofia do córtex motor do cérebro. Porém, vale ressaltar que esta técnica é ineficaz no diagnóstico da doença em pacientes com síndrome bulbar.
Muitos exames laboratoriais são realizados durante o diagnóstico de ELA. Em particular, os médicos podem prescrever medicamentos clínicos e testes bioquímicos sangue, exame do líquido cefalorraquidiano, estudos sorológicos. No entanto, o único método de análise eficaz e confiável ainda é considerado a análise genética molecular. A presença de mutações no gene da superóxido dismutase-1 é considerada suspeita de ELA.
Como os sintomas da esclerose lateral amiotrófica são em muitos aspectos semelhantes às manifestações de outras patologias neurológicas, os médicos devem realizar um diagnóstico diferencial. O diagnóstico mais preciso pode ser feito por meio de ressonância magnética do cérebro e da coluna. Em primeiro lugar, a ELA deve ser diferenciada das doenças musculares, que incluem a miotonia distrófica de Rossolimo-Steinert-Kurshman, a miosite com anomalias celulares e a miodistrofia oculofaríngea.
Também é necessário distinguir a ELA das patologias da medula espinhal:
O diagnóstico diferencial também é necessário para distinguir a doença de patologias sistêmicas, lesões da sinapse neuromuscular e patologias cerebrais, como atrofia de múltiplos sistemas, encefalopatia discirculatória e siringobulbia.
Considera-se que os principais objetivos do tratamento da esclerose lateral amiotrófica são retardar a progressão da doença, bem como eliminar seus sintomas, que pioram significativamente a qualidade de vida do paciente. Deve-se lembrar que a ELA é uma doença grave e incurável que reduz a expectativa de vida de uma pessoa. É por isso que o médico tem o direito de informar o paciente sobre o diagnóstico somente após um exame completo e minucioso.
O tratamento da doença inclui terapia medicamentosa e não medicamentosa. Este último implica medidas de segurança. O paciente deve limitar exercício físico, o que pode acelerar a progressão da ELA. Além disso, é muito importante comer de forma adequada e nutritiva. Terapia medicamentosaé dividido em dois tipos: patogenético e paliativo.
Até o momento, o único medicamento que pode retardar a progressão da ELA é o riluzol. Está comprovado que tomá-lo pode prolongar a vida do paciente em média três meses. Este medicamento é indicado para pacientes com duração da doença inferior a 5 anos. O paciente deve receber 100 mg do medicamento diariamente. Para evitar o risco de hepatite induzida por drogas, a cada três meses é necessário verificar os níveis de AST, ALT e LDH. Como os homens e os fumadores têm concentrações mais baixas de riluzol no sangue, devem limitar o consumo de tabaco ou deixar de fumar completamente. mau hábito. Você precisará tomar o medicamento pelo resto da vida.
Os cientistas tentaram repetidamente usar outros medicamentos para terapia patogenética. No entanto, tais experimentos não se mostraram eficazes. Entre eles estavam:
A eficácia de tomá-lo também não foi comprovada altas doses Cerebrolisina, apesar de este medicamento ser capaz de melhorar ligeiramente o estado dos pacientes.
A terapia paliativa visa eliminar um complexo de sintomas da doença e, assim, melhorar a qualidade de vida do paciente. Para eliminar certos sintomas da ELA, são utilizadas as seguintes técnicas:
Para melhorar o metabolismo muscular, um paciente com ELA pode receber os seguintes medicamentos: creatina, carnitina, solução de levocarnitina, propionato de trimetilhidrazínio. A terapia multivitamínica também é indicada para pacientes, o que envolve o uso de multivitaminas (neuromultivit, milgamma) e ácido tióctico.
Na maioria dos pacientes com ELA, a doença é acompanhada por graves deficiências motoras, incluindo mobilidade limitada. É claro que isso causa grande desconforto ao paciente, que precisa constantemente da ajuda de outras pessoas. As técnicas de correção ortopédica ajudam a eliminar alguns distúrbios do movimento. O médico deve explicar ao paciente que o uso de dispositivos assistivos não indica sua deficiência, mas apenas reduz as dificuldades causadas pela doença.
O sintoma mais fatal da doença é considerado insuficiência respiratória. Seus primeiros sintomas serão fadiga matinal, sonhos vívidos, sonolência diurna e insatisfação com o sono. Para detecção Parada respiratória Numa fase inicial, são realizadas polissonografia e espirografia. Para eliminar a apneia, são indicados medicamentos e ventilação não invasiva. Está comprovado que essas técnicas podem prolongar a vida de um paciente em um ano. Se o paciente necessitar de respiração assistida por mais de 20 horas, o médico levanta a questão de uma transição completa para ventilação invasiva.
Pacientes que tenham sido submetidos a um exame inicial ou a um diagnóstico repetido da doença devem permanecer sob observação ambulatorial. À medida que surgem novos sintomas, eles também devem receber aconselhamento qualificado. Os pacientes devem tomar a maioria dos medicamentos regularmente. Apenas vitaminas e medicamentos miotrópicos são administrados em etapas.
A cada três meses o paciente deve realizar espirografia. Se ele toma riluzol regularmente, ele precisa verificar LDH, AST e ALT a cada seis meses. Se o paciente apresentar disfagia, os níveis de glicose no sangue e o estado trófico devem ser medidos periodicamente. Os pacientes podem escolher o regime de tratamento: podem ficar em casa ou em um hospício.
O prognóstico para pacientes com ELA depende em grande parte do curso da doença. Está comprovado que cerca de 80-90% dos pacientes que apresentam complicações respiratórias graves morrem dentro de 3-5 anos após o aparecimento dos primeiros sinais da doença. Os 10% restantes dos pacientes apresentam um curso benigno da doença. A duração da doença é significativamente reduzida na presença dos seguintes fatores: idade do paciente inferior a 45 anos, início bulbar da ELA, rápida progressão da doença.
A esclerose lateral amiotrófica é doença fatal, afetando as células nervosas do cérebro e da medula espinhal responsáveis pelo movimento. Os neurônios motores (células nervosas que enviam o impulso para a contração muscular) em um paciente com ELA morrem como resultado de uma rápida degeneração, deixando a pessoa paralisada.
Um de pessoas famosas O cientista britânico Stephen Hawking tem esclerose amiotrófica.
Os músculos controlados pelos neurônios motores não têm nutrição adequada e, como resultado, enfraquecem e atrofiam (encolhem). A paralisia ocorre no estágio final da ELA e impede o paciente de funcionar fisicamente, embora permaneça mentalmente intacto. Causas conhecidas Não existem tratamentos ou tratamentos para a esclerose lateral amiotrófica, e a doença pode afetar qualquer pessoa. A causa habitual de morte é a paralisia dos músculos respiratórios que controlam a respiração.
A esclerose lateral amiotrófica é uma doença progressiva do sistema nervoso central.
“A” significa “não”, “mio” significa células musculares e “trófico” refere-se à nutrição. As células nervosas que se estendem do cérebro à medula espinhal (neurônios motores superiores) e da medula espinhal aos nervos periféricos (neurônios motores inferiores) degeneram e morrem inexplicavelmente. "
Lateral" refere-se às áreas da medula espinhal que são afetadas, e "esclerose" ocorre quando tecido duro substitui anteriormente originalmente nervo saudável. As partes do corpo que não são afetadas pela ELA são aquelas que não estão envolvidas no uso dos neurônios motores. A mente permanece muito aguçada e controla a visão, a audição, o olfato, o tato e o paladar. Funções intestinais e Bexiga, via de regra, não são afetados.
A esclerose lateral amiotrófica raramente causa dor, mas os pacientes nos estágios mais avançados da doença dependem de cuidados.
A ELA progride rapidamente e os pacientes paralisados geralmente ficam sob cuidados intensivos em enfermarias ou familiares. Isso pode ter um impacto psicológico devastador nos familiares e no paciente. A maioria dos casos de ELA são fatais dentro de dois a cinco anos, embora aproximadamente 10% sobrevivam oito anos ou mais.
A esclerose lateral amiotrófica não é uma doença rara. Afecta aproximadamente sete pessoas em cada 100 000. A maioria das pessoas com ELA tem entre 40 e 70 anos de idade. Aproximadamente 5-10% dos casos apresentam um padrão de hereditariedade.

Em 1991, uma equipe de pesquisadores ligou o ALSD familiar ao cromossomo 21. Em 1993, foi descoberto que o cromossomo 21 tinha defeitos estruturais no gene SOD1 (superóxido dismutase).
O gene SOD1 é uma enzima que protege os neurônios motores dos radicais livres. A ilha de Guam, a Nova Guiné Ocidental e a península japonesa apresentam altas taxas de ELA, levando alguns teóricos a acreditar que a composição genética pode ser suscetível a razões ambientais, como níveis altos níveis de mercúrio e chumbo nessas áreas.

O padrão de herança é autossômico dominante, o que significa que os filhos de um pai afetado têm 50% de chance de herdar o distúrbio. Na maioria dos casos, é devido a uma mutação genética esporádica, o que significa que a mutação ocorre apenas na pessoa afetada. Acredita-se que mutações esporádicas resultem de causas biológicas e ambientais.
EM em casos raros uma mutação é evidente no NFH, o gene que codifica o neurofilamento (uma estrutura que mantém a forma da célula). A esclerose lateral amiotrópica familiar tem sido associada a outras localizações cromossômicas, mas os genes exatos não foram identificados. A Universidade Thomas Jefferson, na Filadélfia, aprovou recentemente um projeto terapia de genes BAIXO.
O projeto visa injetar um vírus adeno-associado que transporta uma cópia normal do gene EAAT2 na medula espinhal de um paciente com ELA, onde os neurônios motores morrem.

A esclerose lateral amiotrófica afeta todas as pessoas e tanto homens como mulheres correm o mesmo risco. A ELA pode ocorrer em qualquer idade e as chances de desenvolvê-la aumentam com a idade. Foram relatados casos de adolescentes com ELA. Uma pessoa só precisa herdar um gene defeituoso de um dos pais para causar a doença.
A doença começa lentamente, afetando apenas um membro, como um braço ou uma perna, e gradualmente se espalha para mais membros e músculos. Quando os músculos não têm a nutrição adequada de que necessitam, eles começam a ficar finos e a deteriorar-se. Esta condição é um sintoma de esclerose lateral amiotrófica.
A perda muscular é causada pela incapacidade dos neurônios motores em degeneração de enviar sinais aos músculos que lhes permitem funcionar e crescer. Exemplos típicos os sintomas da ELA são cãibras musculares e espasmos, fraqueza nos braços, pernas ou tornozelos, dificuldade para falar e engolir. Outros sintomas iniciais incluem rigidez nos braços e pernas, perda de uma perna, perda de peso, fadiga e dificuldade de expressão facial.

Um dos mais primeiros sintomas ALS é fraqueza nos músculos bulbares. Esses músculos da boca e da garganta ajudam na mastigação, na deglutição e na fala. A fraqueza nesses grupos musculares geralmente causa problemas como fala arrastada, dificuldade para falar e rouquidão. Outro sintoma da ELA que geralmente ocorre após o aparecimento dos primeiros sintomas são espasmos musculares persistentes (fasciculação).
A fasciculação quase nunca é o primeiro sinal de ELA.
À medida que a doença progride, os músculos respiratórios (músculos respiratórios) enfraquecem, levando ao aumento da dificuldade em respirar, tossir e possivelmente inalar alimentos ou saliva. O potencial de infecção pulmonar aumenta e pode levar à morte.
A comunicação se torna muito difícil. Uma maneira de conseguir opinião com outras pessoas é usar os olhos.
Piscar é um dos modos que os pacientes com esclerose lateral amiotrófica serão forçados a usar para continuar a comunicação. À medida que a doença progride, as vítimas perdem gradualmente o uso das pernas, braços, pernas e músculos do pescoço, e a paralisia resulta nos grupos musculares afetados. Eles só conseguem falar e engolir com grande dificuldade.
A função sexual não é afetada.
A respiração torna-se cada vez mais difícil e os pacientes com ELA podem optar por prolongar a vida com ventilação assistida, o que pode reduzir o risco de morte por infecções como a pneumonia.

ALS é difícil de diagnosticar. Uma série de testes de diagnóstico descartarão a entrada e a exclusão de outros razões possíveis e doenças que se assemelham à ELA. Testes de eletrodiagnóstico, como eletromiografia (EMG) e velocidade de condução nervosa, são usados para diagnosticar ELA.
São realizados exames de sangue e urina, biópsias musculares e/ou nervosas, além de ressonância magnética (RM), mielogramas da coluna cervical e exames completos. exame neurológico Implementação do programa fisioterapia, provisão cadeira de rodas ou andadores, a assistência durante o banho auxilia o paciente com ELA. Outras considerações incluem o fornecimento de produtos fáceis de engolir, cuidados com a pele e apoio emocional.
Entre as doenças neurológicas existem aquelas que podem ser tratadas com sucesso. Mas também há aqueles que só podem ser suspensos - pertencem ao grupo dos distróficos degenerativos. Uma dessas doenças é BAIXO- esclerose lateral amiotrófica. Leia sobre outros aqui.
No esclerose lateral amiotrófica a morte gradual ocorre células nervosas, localizado nos cornos laterais da medula espinhal e responsável pela função motora.
Ao mesmo tempo, vários distúrbios motores se desenvolvem. A doença progride gradualmente e, finalmente, leva a resultado fatal devido a danos nos músculos respiratórios.
ELA é suficiente doença rara. Não há mais pessoas no mundo que entendam três pessoas entre cem mil. Com menos de 65 anos, a doença é uma vez e meia mais comum em homens, depois homens e mulheres são igualmente afetados.
surgir esclerose lateral amiotrófica Pode ser em qualquer idade - desde adolescentes até idosos. Em dez por cento dos casos isso formulário familiar doenças. Os restantes casos são esporádicos.
Existem várias formas de ELA, dependendo da primeira parte da medula espinhal afetada:
Não existe uma causa única para o desenvolvimento da esclerose lateral amiotrófica; ela ocorre como resultado de uma combinação de vários fatores.
Os fatores causais podem vir do próprio corpo e do ambiente externo:
Os sintomas da esclerose lateral amiotrófica variam dependendo do nível de dano:
Existe uma forma bulbar - com ela a doença começa com os seguintes sintomas:

A variante mais desfavorável do desenvolvimento da esclerose lateral amiotrófica é a forma generalizada primária:
ELA - doença grave com o inevitável fatal. Portanto, para fazer tal diagnóstico é necessário realizar muitas pesquisas.
O que é levado em consideração ao fazer um diagnóstico:
Nesse caso, desenvolvem-se paresia e atrofia dos músculos das partes do corpo que são inervadas pelos neurônios afetados:
Aparecem reflexos fortalecidos, sinais piramidais e espasticidade:
Um diagnóstico confiável de esclerose lateral amiotrófica é feito quando os neurônios motores periféricos e centrais são danificados em três partes da medula espinhal.
Qual métodos instrumentais realizado para diagnosticar ELA:
De métodos laboratoriais a única coisa específica é mapeamento genético— identificação dos genes responsáveis pelo desenvolvimento da esclerose lateral amiotrófica.
O diagnóstico diferencial deve ser realizado para excluir todas essas doenças:
A esclerose lateral amiotrófica não pode ser tratada. O único resultado desta doença é a morte por insuficiência respiratória aguda.
Porém, o tratamento ainda é utilizado e tem dois objetivos:
Para atingir o primeiro objetivo, apenas um medicamento é utilizado - o Riluzol. Prolonga a vida dos pacientes com esclerose lateral amiotrófica em três meses.
Na maioria dos casos, apenas o tratamento sintomático é utilizado:
No distúrbios de postura e são prescritos deformidades nos pés, espartilhos e sapatos ortopédicos. Para prevenção trombose — meias de compressão ou curativo bandagem elástica. Se o processo for interrompido engolir- purê de alimentos, alimentação por sonda nasogástrica.
Atualização: dezembro de 2018
A esclerose lateral amiotrófica, ou doença de Lou Gehrig, é uma doença rapidamente progressiva do sistema nervoso, caracterizada por danos aos neurônios motores da medula espinhal, do córtex e do tronco cerebral. Além disso, os ramos motores dos neurônios cranianos (trigêmeo, facial, glossofaríngeo) estão envolvidos no processo patológico.
A doença é extremamente rara, aproximadamente 2 a 5 pessoas por 100.000. Acredita-se que homens com mais de 50 anos sejam afetados com mais frequência. A doença de Lou Gehrig não abre exceções para ninguém: afeta pessoas de diferentes classes sociais e de diversas profissões (atores, senadores, ganhadores do Nobel, engenheiros, professores). O paciente mais famoso foi o campeão mundial de beisebol Loi Gering, que deu nome à doença.
Na Rússia, a esclerose lateral amiotrófica tornou-se generalizada. Atualmente, o número de doentes é de aproximadamente 15.000-20.000 na população. Entre as pessoas famosas da Rússia que esta patologia, podemos citar o compositor Dmitry Shostakovich, o político Yuri Gladkov e o cantor pop Vladimir Migulya.
A doença se baseia no acúmulo de proteínas patológicas insolúveis nas células motoras do sistema nervoso, levando à sua morte. A causa da doença é atualmente desconhecida, mas existem muitas teorias. As principais teorias incluem:
Qualquer forma da doença tem o mesmo início: os pacientes queixam-se de aumento fraqueza muscular, diminuição da massa muscular e aparecimento de fasciculações (espasmos musculares).
Forma bulbar de ELA
caracterizado por sintomas de danos aos nervos cranianos (9, 10 e 12 pares):Variante cervicotorácica A doença atinge principalmente os membros superiores do paciente, simetricamente em ambos os lados:
Forma lombossacra geralmente começa com uma sensação de fraqueza nas extremidades inferiores.
Independentemente de qual variante predomine nos pacientes no início da doença, o desfecho ainda é o mesmo. A doença progride de forma constante, espalhando-se por todos os músculos do corpo, incluindo os músculos respiratórios. Quando os músculos respiratórios falham, o paciente começa a precisar ventilação artificial pulmões e cuidados constantes.
Na minha prática, observei dois pacientes com ELA, um homem e uma mulher. Eles se distinguiam pela cor do cabelo ruivo e pela idade relativamente jovem (até 40 anos). Externamente eram muito parecidos: não havia nenhum sinal de músculos, um rosto amigável e a boca sempre ligeiramente aberta.
Esses pacientes morrem na maioria dos casos de doenças concomitantes(pneumonia, sepse). Mesmo com os devidos cuidados, desenvolvem escaras (ver), pneumonia hipostática. Percebendo a gravidade de sua doença, os pacientes caem em depressão, apatia e deixam de se interessar pelo mundo exterior e por seus entes queridos.
Com o tempo, a psique do paciente sofre fortes mudanças. O paciente que observei durante um ano se distinguiu por seus caprichos, labilidade emocional, agressividade e falta de controle. Os testes de inteligência mostraram um declínio em seu pensamento, habilidades mentais, memória, atenção.
Os principais métodos de diagnóstico incluem:
O que acontece na ELA
Pelo fato da ELA apresentar sintomas semelhantes a outras doenças, é feito diagnóstico diferencial:
O tratamento da doença é atualmente ineficaz. Medicamentos e o cuidado adequado ao paciente apenas prolonga a expectativa de vida sem proporcionar recuperação total. A terapia sintomática inclui:
A terapia complexa inclui necessariamente alimentação por sonda nasogástrica, massagem, terapia por exercícios com médico e consultas com psicólogo.
Infelizmente, o prognóstico para a esclerose lateral amiotrófica é desfavorável. Os pacientes morrem literalmente dentro de alguns meses ou anos, duração média vida em pacientes:
Prognóstico mais favorável para casos hereditários da doença associados a mutações no gene da superóxido dismutase-1.
A situação na Rússia é ofuscada pelo facto de os pacientes não receberem cuidados adequados, como evidenciado pelo facto de o Riluzote, um medicamento que retarda o curso da doença, só ter sido registado na Rússia em 2011, e apenas no mesmo ano, a própria doença foi incluída na lista de "raras". Mas em Moscou existem:
Por fim, gostaria de acrescentar sobre o evento de caridade Ice Bucket Challenge que aconteceu em julho de 2014. O objetivo era arrecadar fundos para apoiar pacientes com esclerose lateral amiotrófica e se tornou bastante difundido. Os organizadores conseguiram arrecadar mais de US$ 40 milhões.
A essência da ação era que uma pessoa se encharcava com um balde de água gelada e capturava em vídeo ou doava uma certa quantia em dinheiro para uma organização de caridade. O evento tornou-se bastante popular devido à participação de artistas populares, atores e até políticos.

O que seria do Ano Novo sem champanhe, tangerinas, Olivier, formol e o “arenque com casaco de pele” preferido de todos. Com o último...

Vamos preparar os ingredientes necessários para os biscoitos. A primeira coisa a fazer é colocar a água para ferver. Nós precisamos...

É possível cadastrar funcionário para o cargo de diretor financeiro - contador-chefe? O contador-chefe afirma que sim, mas...

O chefe de uma pequena empresa pode gerenciar facilmente o orçamento de forma independente. VERIFICADO! Se você está fazendo um orçamento...

A criação de novos projetos envolve uma justificação económica preliminar para a sua viabilidade, posterior...
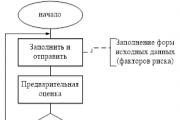
O reporte é gerado pelo RM, é acordado (aprovado) pelo Comité de Risco no âmbito do Conselho de Administração e transmitido a...

Na beira de um prado grande, muito grande, em uma longa folha de grama esmeralda vivia uma pequena joaninha. O pequeno de Deus...

Hoje em dia é bastante comum as pessoas se voltarem para as estrelas. Com a ajuda de um horóscopo uma pessoa pode descobrir...

Negócios ou amigável. Se você estava perseguindo um estranho, isso significa o seu nível de confiança no sexo feminino...

de acordo com o livro dos sonhos de Freud Se você sonhou que estava pescando, isso significa que na vida real dificilmente você consegue desligar...

O turbilhão do Ano Novo nos girará tão rápida e rapidamente que é hora de pensar cuidadosamente sobre o Ano Novo...

O artigo oferece algumas das mais deliciosas receitas para fazer vinagrete. Vinagrete é uma salada deliciosa...

4 de julho de 2015, 20h33 Ultimamente nossa Nastena tem desistido completamente dos doces (e graças a Deus), mas...

Bolo de chocolate perfumado e muito saboroso, é impossível parar de comer! O bolo Beijo do Negro é muito fácil de preparar,...

Vamos preparar os ingredientes necessários para os biscoitos. A primeira coisa a fazer é colocar a água para ferver. Nós...

É possível cadastrar funcionário para o cargo de diretor financeiro - contador-chefe? O contador-chefe afirma...