Interpretação das cartas de tarô: o laço do Diabo e seu significado no layout
Quantas vezes vemos esse monstro com chifres de cabra quando distribuímos cartas de Tarô. "Diabo" é a personificação do inferno e da morte...
Eritremia ( policitemia vera, doença de Váquez) – Doença hereditária sistema sanguíneo, ocorrendo predominantemente em mulheres idosas.
Aumento da contagem de glóbulos vermelhos na policitemia vera
Esta patologia é caracterizada por hipertrofia maligna medula óssea. Na maioria das vezes, esta patologia é conhecida pelos pacientes como câncer de sangue (embora tal julgamento seja errôneo) e leva a um aumento progressivo no número de células sanguíneas, principalmente glóbulos vermelhos (o número de outros elementos também aumenta). Como resultado do aumento do seu número, observa-se um aumento do hematócrito, o que leva à diminuição das propriedades reológicas do sangue, à diminuição da velocidade do fluxo sanguíneo através dos vasos e, como consequência, ao aumento da formação de trombos e deterioração no fornecimento de tecidos.
Estas razões levam ao fato de que a maioria dos tecidos experimenta fome de oxigênio, o que reduz sua atividade funcional (síndrome isquêmica). A policitemia vera ocorre principalmente em mulheres. Os homens adoecem com menos frequência; a incidência desta patologia é de aproximadamente 3:2.
Em média, a doença de Vaquez ocorre por volta dos 40 anos de idade, com pico de sintomas entre 60 e 70 anos de idade. Rastreável predisposição hereditáriaà doença. Na população, a eritremia é bastante rara - cerca de 30 casos por milhão de habitantes.

A eritremia é uma saturação excessiva do sangue com glóbulos vermelhos, o que leva a vários distúrbios teciduais e vasculares. Entre os sintomas mais comuns estão:
Entre sintomas comuns, se ocorrer eritremia, o primeiro lugar vem dor de cabeça, tonturas, sensação de peso na cabeça, zumbido, síndrome de fraqueza geral (todos os sintomas são causados por diminuição da oxigenação dos tecidos, circulação sanguínea prejudicada em certas partes do corpo). No diagnóstico, não são utilizados como critério obrigatório, pois podem corresponder a qualquer doença sistêmica.
A policitemia vera ocorre em três estágios (fases):
Todas as etapas ocorrem sequencialmente, e o diagnóstico da doença (exame de sangue) torna-se informativo a partir da fase dos sinais clínicos.

Para fazer um diagnóstico, um exame de sangue geral desempenha um papel decisivo. Apresenta eritrocitose pronunciada, aumento dos níveis de hemoglobina e. O mais confiável é a análise do puntiforme da medula óssea, que revela sinais de hiperplasia do germe eritróide, e também calcula quantas células nele estão presentes e qual a sua distribuição morfológica.
Para esclarecer a natureza da patologia concomitante, recomenda-se a realização de uma análise bioquímica, que fornece informações sobre o estado do fígado e dos rins. No caso de trombose maciça, o estado dos fatores de coagulação sanguínea é avaliado através da análise de sua coagulabilidade - um coagulograma.
Outros estudos (ultrassonografia, tomografia computadorizada, ressonância magnética) fornecem apenas uma ideia indireta do estado do corpo e não são utilizados para fazer um diagnóstico.
Apesar da variedade e gravidade das manifestações da doença de Vaquez, existem relativamente poucos tratamentos para ela. Depende do que a análise do hemograma mostrou, se houve desenvolvimento de síndrome citológica e quais sintomas o paciente apresenta.
Como mencionado acima, a doença é causada por um aumento da concentração de células sanguíneas (especialmente glóbulos vermelhos), que se desenvolve devido à hiperplasia da medula óssea. Devido a isso, análise correta As formas de desenvolvimento da doença permitem determinar os princípios básicos do tratamento patogenético, que incluem a redução do número de células sanguíneas e a atuação direta nos locais de sua formação. Isto é conseguido através dos seguintes métodos de tratamento:

Tal tratamento deve ser acompanhado da prescrição de medicamentos antiplaquetários como aspirina, carrilhão, clopidogrel ou anticoagulantes (heparina). O uso desses medicamentos com um dos procedimentos aumenta significativamente a eficácia da terapia do que usá-los separadamente.
Também é recomendado adicionar alguns medicamentos citostáticos ao regime de tratamento (se a causa da hiperplasia da medula óssea for câncer), interferons (se secundários complicações virais) ou hormônios (são usadas principalmente dexametasona e prednisolona), que podem melhorar o prognóstico da doença.

Todas as complicações da doença são causadas pelo desenvolvimento de trombose vascular. Como resultado de seu bloqueio, podem ocorrer infartos de órgãos internos (coração, fígado, baço, cérebro), aterosclerose obliterante (quando trombose de vasos afetados por placas ateroscleróticas). membros inferiores). O excesso de hemoglobina no sangue provoca o desenvolvimento de hemocromatose, urolitíase ou gota.
Todos eles se desenvolvem secundariamente e requerem a eliminação da causa subjacente - a eritrocitose, para um tratamento mais eficaz.
Quanto ao prognóstico da doença, muito depende da idade em que o tratamento foi iniciado, quais métodos foram utilizados e se foram eficazes.
Conforme mencionado no início, a policitemia vera tende a se desenvolver mais tarde. Se for observado o aparecimento dos principais sintomas em jovens (25 a 40 anos), a doença é maligna, ou seja, o prognóstico é desfavorável e as complicações secundárias evoluem muito mais rapidamente. Assim, quanto mais tarde for observado o desenvolvimento da doença, mais benigna ela será. Quando medicamentos prescritos adequadamente são usados, a expectativa de vida dos pacientes melhora significativamente. Esses pacientes podem viver normalmente com sua doença por muito tempo (até várias décadas).
Respondendo à pergunta sobre qual poderia ser o resultado da eritremia, deve-se notar que tudo depende de:
Na maioria das vezes, devido a danos no fígado e no baço, a policitemia transita para forma crônica. A vida útil permanece quase a mesma, e com seleção correta medicamentos podem chegar a dezenas de anos (prognóstico em relação
Entre as doenças do sangue, há muitas que causam diminuição de vários elementos - glóbulos vermelhos, glóbulos brancos, plaquetas. Mas em algumas patologias, ao contrário, ocorre um aumento descontrolado do número de células sanguíneas. Condição em que há um aumento crônico no número de glóbulos vermelhos e outros alterações patológicas, chamada “policitemia vera”.
A policitemia primária (verdadeira) é uma doença do sangue do grupo das leucemias que ocorre de forma idiopática (sem razões visíveis), prossegue por muito tempo (cronicamente) e é caracterizado por aumento do número de glóbulos vermelhos, aumento do hematócrito e da viscosidade do sangue. Os sinônimos para o nome da patologia são doença de Vaquez-Osler, eritremia, eritrocitose primária. As consequências da eritrocitose e do espessamento do sangue nesta doença mieloproliferativa podem ser graves e estão relacionadas com o risco de trombose, aumento do tamanho e ruptura do baço, aumento do volume de sangue circulante, etc.
A eritremia é considerada um processo tumoral maligno, causado pelo aumento da proliferação (hiperplasia) das células da medula óssea. O processo patológico é especialmente forte no germe eritroblástico, uma parte da medula óssea que consiste em eritroblastos e normoblastos. A patogênese das principais manifestações está associada ao aparecimento de um grande número de glóbulos vermelhos no sangue, bem como a um ligeiro aumento no número de plaquetas e neutrófilos ( leucócitos neutrófilos). As células sanguíneas são morfologicamente normais, mas seu número é anormal. Como resultado, a viscosidade do sangue e a quantidade de sangue na corrente sanguínea circulante aumentam. O resultado é um fluxo sanguíneo mais lento, a formação de coágulos sanguíneos, a interrupção do suprimento sanguíneo local aos tecidos e sua hipóxia.
Se inicialmente o paciente apresenta eritrocitose primária com mais frequência, ou seja, apenas o número de glóbulos vermelhos aumenta, então outras alterações começam a afetar outras células sanguíneas. Hematopoiese extramedular ( formação patológica sangue fora da medula óssea) ocorre nos órgãos do peritônio - no fígado e no baço, onde parte da eritropoiese - processo de formação dos glóbulos vermelhos - também está localizada. Numa fase posterior da doença, é encurtado vida útil eritrócitos, anemia, trombocitopenia, mielofibrose podem se desenvolver e as células precursoras de leucócitos e eritrócitos entram na corrente sanguínea geral sem amadurecer. Em aproximadamente 10% dos casos, a patologia evolui para leucemia aguda.
O estudo e a primeira descrição da eritrocitose foram feitos em 1892 por Vaquez, e em 1903 o cientista Osler sugeriu que a causa da doença era um mau funcionamento da medula óssea. A policitemia vera é observada com mais frequência do que outras patologias semelhantes, mas ainda é bastante rara. É diagnosticado em aproximadamente 5 pessoas por ano por 1 milhão de habitantes. Na maioria das vezes, a doença ocorre em pessoas com mais de 50 anos de idade, idade Média detecção - 60 anos. Em crianças, esse diagnóstico é feito muito raramente, principalmente após os 12 anos. Em média, apenas 5% dos casos têm menos de 40 anos de idade. Os homens sofrem desta patologia com mais frequência do que as mulheres. Na estrutura geral das doenças mieloproliferativas crônicas, a policitemia vera ocupa o 4º lugar. Às vezes é herdado, então há casos familiares.
A forma primária da doença é considerada hereditária e é transmitida de forma autossômica recessiva. Neste caso, é frequentemente referida como “policitemia familiar”. Mas na maioria das vezes a eritremia é uma condição secundária, representando uma das manifestações de um processo patológico geral. As causas exatas não foram estabelecidas, mas existem várias teorias sobre o aparecimento da policitemia vera. Assim, existe uma ligação entre o desenvolvimento da doença e a transformação das células-tronco, quando ocorre uma mutação da tirosina quinase, que ocorre na policitemia vera com mais frequência do que em outras doenças do sangue.
Estudos de células na eritremia revelaram a origem clonal da patologia em muitos pacientes, uma vez que a mesma enzima foi detectada em leucócitos, plaquetas e eritrócitos. A teoria clonal também é confirmada por estudos citológicos em andamento sobre o cariótipo dos grupos cromossômicos, onde foram identificados vários defeitos, semelhantes em diferentes pacientes. Existe também uma teoria genético-viral, segundo a qual até 15 tipos de vírus podem invadir o corpo e, com a participação de uma série de fatores provocadores, levar ao mau funcionamento da medula óssea. Eles penetram nos precursores das células sanguíneas, que então, em vez de amadurecerem normalmente, começam a se dividir e a formar novos glóbulos vermelhos e outras células.
Quanto aos fatores de risco para o desenvolvimento de policitemia vera, presumivelmente podem ser os seguintes:
Assim, a principal causa da eritrocitose secundária são todas as condições que de uma forma ou de outra provocam hipóxia tecidual, estresse ao organismo ou sua intoxicação. Além do mais, grande influência pode ter um efeito no cérebro e na produção de células sanguíneas adicionais processos oncológicos, patologias endócrinas, doenças hepáticas.

A doença é classificada nas seguintes fases:
Muitas vezes esta patologia é assintomática, mas apenas nos estágios iniciais. Posteriormente, a doença do paciente se manifesta de uma forma ou de outra, e os sintomas específicos podem ser variados. Basicamente, o complexo de sintomas inclui os seguintes sinais principais:
Existem outros sinais de policitemia vera dos quais uma pessoa pode reclamar, mas não são muito específicos e podem ser característicos de diferentes patologias:
Em geral, a doença é caracterizada por um curso longo e às vezes benigno, principalmente com tratamento adequado. Mas algumas pessoas, especialmente aquelas que não recebem terapia, podem apresentar sintomas de início precoce várias consequências policitemia vera.

Na maioria das vezes, as complicações estão associadas à trombose e embolia das veias e vasos do baço, fígado, pernas, cérebro e outras áreas do corpo. Isto leva a consequências diferentes dependendo do tamanho do coágulo sanguíneo e da área afetada. Podem ocorrer ataques isquêmicos transitórios, acidentes vasculares cerebrais, tromboflebite e flebotrombose de veias superficiais e profundas, bloqueio de vasos retinianos e cegueira, infarto de órgãos internos e infarto do miocárdio.
Nos estágios mais avançados da patologia, muitas vezes aparecem cálculos renais ( doença de urolitíase), gota, nefroesclerose, cirrose hepática. É provável que ocorram complicações devido ao sangramento dos tecidos - sangramento de úlceras gastrointestinais, anemia. Por parte do coração, além do infarto do miocárdio, também são possíveis sinais de miocardiosclerose e insuficiência cardíaca. Existe também a possibilidade de transição da policitemia vera para leucemia aguda, leucemia crônica e outras oncopatologias.
Fazer o diagnóstico desta doença não é fácil, principalmente na ausência de quadro clínico característico e na presença apenas de sintomas gerais. No entanto, a totalidade dos dados hematológicos e testes bioquímicos, bem como algumas características distintivas da aparência do paciente, juntamente com suas queixas, ajudarão o médico a determinar a causa das alterações ocorridas.
Os principais indicadores para estabelecer o diagnóstico de policitemia vera são: análise geral sangue - o número de glóbulos vermelhos e hematócrito. Nos homens, pode-se suspeitar do desenvolvimento desta doença se o número de glóbulos vermelhos for superior a 5,7*10*9/l, a hemoglobina for superior a 177 g/l e o hematócrito estiver acima de 52%. Nas mulheres, os valores excessivos são observados se forem superiores a 5,2*10*9/l, 172 g/l, 48-50%, respectivamente. Os números indicados são típicos para estágios iniciais patologia e, à medida que se desenvolve, tornam-se ainda maiores. Além disso, é importante avaliar a massa de hemácias circulantes, que normalmente é de até 36 ml/kg para homens e de até 32 ml/kg para mulheres.
Outros parâmetros sanguíneos (bioquímica, análise geral e outros exames), que, em combinação com os distúrbios descritos e em combinação entre si, refletem o quadro do desenvolvimento da eritrocitose primária ou secundária:
Na fase da mielofibrose, os níveis de hemoglobina e glóbulos vermelhos podem voltar ao normal, mas ao mesmo tempo o número de leucócitos aumenta muito, aparecem suas formas imaturas e é diagnosticada a presença de eritroblastos. Quanto ao mielograma, obtido por punção da medula óssea, são reveladas as seguintes alterações:
Existem outros critérios pelos quais o médico pode tirar uma conclusão sobre as alterações que ocorrem e são características da policitemia vera:
Todos os critérios descritos acima, exceto os três principais, que são considerados grandes, são pequenos. Quanto aos principais critérios diagnósticos, são aumento da massa de glóbulos vermelhos circulantes, esplenomegalia, supersaturação Sangue arterial oxigênio. Para fazer um diagnóstico, geralmente é suficiente ter três destes critérios maiores, que são combinados com dois ou três menores. Diagnóstico diferencial realizado por um hematologista entre condições acompanhadas de eritrocitose - cardiopatias, tuberculose, tumores, etc.

Como antigamente homem procurar ajuda, mais eficaz a terapia pode ser. No terceiro estágio, ou quando outro processo tumoral se sobrepõe à eritremia, a terapia sintomática é realizada em combinação com o tratamento com quimioterapia. O tratamento quimioterápico pode ser recomendado em outras fases da doença, mas nem sempre o organismo responde adequadamente. Dentre os remédios sintomáticos que melhoram a qualidade de vida, são utilizados:
Os métodos de tratamento para policitemia vera podem incluir:
O transplante de medula óssea para uma doença como a eritremia geralmente leva a resultados desfavoráveis e, portanto, é raramente utilizado. Em alguns casos, a esplenectomia está indicada, mas com o desenvolvimento de leucemia aguda, tal operação não é realizada mesmo com esplenomegalia grave.
Durante a gravidez, esta patologia ocorre raramente. Porém, se houver predisposição (hereditária ou de fatores secundários), a gravidez, o parto e o aborto podem se tornar um gatilho para o desenvolvimento da policitemia vera. A gravidez sempre piora o curso da doença e seu desfecho pode ser mais grave do que fora da gestação. No entanto, em 50% dos casos, a gravidez termina num parto bem sucedido. A metade restante se deve a abortos espontâneos, atrasos no desenvolvimento e anomalias estruturais do feto.
O tratamento da doença em gestantes não é fácil. A maioria dos medicamentos é estritamente contra-indicada, pois possuem uma propriedade teratogênica pronunciada. Portanto, durante a gravidez, a terapia é realizada predominantemente com sangria e, se necessário, glicocorticosteróides. Para prevenir complicações e detecção precoce doenças em gestantes, os exames de sangue devem ser realizados regularmente de acordo com cronograma designado pelo obstetra-ginecologista supervisor.
É estritamente proibido o uso de diuréticos, que engrossam ainda mais o sangue. Também em nossa época, o uso de preparações de fósforo radioativo, que inibem gravemente a mielopoiese e muitas vezes levam ao desenvolvimento de leucemia, é limitado. Também não é possível manter o mesmo sistema nutricional: a dieta deve mudar. Todos os alimentos que melhoram a hematopoiese, como o fígado, são proibidos. É melhor criar uma dieta de vegetais lácteos e evitar o excesso de carne.
O paciente não deve sobrecarregar o corpo, praticar esportes extenuantes ou ignorar o descanso regular. O tratamento com remédios populares pode ser utilizado, mas somente após o médico ter estudado cuidadosamente todos os remédios de acordo com sua composição, para evitar o aumento da produção de glóbulos vermelhos. Na maioria das vezes, a terapia sintomática é usada para remover o ácido úrico, reduzir a dor e a coceira na pele, etc.
Os métodos de prevenção ainda não foram desenvolvidos. O prognóstico para a vida varia dependendo da gravidade da doença. Sem tratamento, até um terço dos pacientes morre nos primeiros 5 anos após o diagnóstico. Se você realizar uma terapia completa, poderá prolongar a vida de uma pessoa para 10 a 15 anos ou mais. A causa mais comum de morte é a trombose, e apenas ocasionalmente as pessoas morrem de câncer no sangue (leucemia) ou sangramento grave.
Policitemia é doença crônica, em que o conteúdo dos eritrócitos (vermelho células sanguíneas) em sangue. Além disso, com esta patologia, em 70% dos pacientes, o número de plaquetas e leucócitos aumenta.
A doença não é muito comum - não são registrados mais de cinco casos anualmente por um milhão de pessoas. Na maioria das vezes, a doença policitemia se desenvolve em pessoas de meia-idade e idosos. Segundo as estatísticas, os homens sofrem desta patologia cinco vezes mais do que as mulheres. Hoje examinaremos mais de perto uma condição como a policitemia, os sintomas e o tratamento da patologia serão descritos a seguir.
A policitemia não é doença maligna. Até o momento, as causas exatas da doença são desconhecidas. Acredita-se que o desenvolvimento da patologia seja causado por uma mutação de uma enzima especial na medula óssea. As alterações genéticas levam à divisão e crescimento excessivos de todas as células sanguíneas, especialmente dos glóbulos vermelhos.
Existem dois grupos da doença:
Policitemia verdadeira, ou doença de Vaquez, que por sua vez é dividida em primária (ou seja, atua como uma doença independente) e secundária (a policitemia secundária se desenvolve devido a doenças pulmonares crônicas, tumores, hidronefrose e elevação).
Policitemia relativa (estresse ou falsa) - com este estado o nível de glóbulos vermelhos permanece dentro dos limites normais.
Muitas vezes a doença é assintomática. Às vezes, como resultado de um exame por razões completamente diferentes, a policitemia vera pode ser detectada acidentalmente. Consideraremos os sintomas aos quais você deve prestar atenção a seguir.
Com policitemia na pele, mais frequentemente na região do pescoço, dilatada veias safenas. Com esta patologia, a pele adquire uma cor cereja avermelhada, especialmente perceptível em áreas abertas do corpo - pescoço, mãos, rosto. A membrana mucosa dos lábios e da língua é de cor vermelho-azulada, a parte branca dos olhos parece injetada de sangue.
Tais alterações são causadas pelo transbordamento de sangue, rico em glóbulos vermelhos, de todos os vasos superficiais e pela desaceleração de suas propriedades reológicas (velocidade de movimento), como resultado da qual a parte principal da hemoglobina (pigmento vermelho) passa para um forma reduzida (isto é, sofre alterações químicas) e muda de cor.
Quase metade dos pacientes que sofrem de policitemia apresentam sintomas graves comichão na pele, especialmente perceptível após um banho quente. Esse fenômeno atua como um sinal específico de policitemia vera. A coceira ocorre devido à liberação substâncias ativas no sangue, em particular a histamina, que é capaz de expandir os capilares da pele, o que leva ao aumento da circulação sanguínea nos mesmos e ao aparecimento de sensações específicas.

Este fenômeno é caracterizado por fortes dores de curta duração na área das pontas dos dedos. São provocadas pelo aumento do nível de plaquetas nos pequenos vasos da mão, resultando na formação de numerosos microtrombos, obstruindo as arteríolas e bloqueando o fluxo de sangue para os tecidos dos dedos. Sinais externos Essa condição é a vermelhidão e o aparecimento de manchas azuladas na pele. Para tanto, recomenda-se tomar aspirina.
Além do baço, o fígado, ou melhor, seu tamanho, também pode mudar. Esses órgãos estão diretamente envolvidos na formação e destruição das células sanguíneas. Um aumento na concentração deste último leva a um aumento no tamanho do fígado e do baço.
Tão sério patologia cirúrgica desenvolve-se devido à trombose de pequenos vasos da membrana mucosa trato digestivo. O resultado agudo é a necrose (necrose) de uma seção da parede do órgão e a formação de um defeito ulcerativo em seu lugar. Além disso, a resistência do estômago ao Helicobacter (um microrganismo que causa gastrite e úlceras) é reduzida.

As veias das extremidades inferiores são mais suscetíveis a esta patologia. da parede do vaso, pode, contornando o coração, penetrar na circulação pulmonar (pulmões) e provocar EP (tromboembolismo artéria pulmonar) - uma condição incompatível com a vida.
Apesar de o número de plaquetas no sangue periférico mudar e sua coagulabilidade aumentar, pode ocorrer sangramento gengival com policitemia.
Quando o nível de ácido úrico aumenta, seus sais se depositam em várias articulações e provocam uma síndrome de dor aguda.
Com uma doença como a policitemia, os sintomas podem ser semelhantes aos sinais de outras patologias (por exemplo, anemia): dores de cabeça, fadiga constante, zumbido, tontura, "arrepios" diante dos olhos, falta de ar. Um aumento nas propriedades de viscosidade do sangue ativa a reação compensatória dos vasos sanguíneos, como resultado, um aumento na pressão arterial. Com esta patologia, são frequentemente observadas complicações na forma de insuficiência cardíaca e microcardiosclerose (substituição tecido muscular coração conectivo, preenchendo o defeito, mas não desempenhando as funções necessárias).

A policitemia é detectada pelos resultados de um exame de sangue geral, que revela:
aumento do número de hemácias de 6,5 para 7,5,10^12/l;
aumento do nível de hemoglobina - até 240 g/l;
o volume total de eritrócitos (TEV) excede 52%.
Uma vez que o número de glóbulos vermelhos não pode ser calculado com base nas medições dos valores acima, diagnóstico de radionuclídeos. Se a massa de glóbulos vermelhos exceder 36 ml/kg nos homens e 32 ml/kg nas mulheres, isso indica de forma confiável a presença da doença de Vaquez.

Na policitemia, a morfologia das hemácias é preservada, ou seja, não alteram sua forma e tamanho normais. No entanto, com o desenvolvimento de anemia como resultado de aumento de sangramento ou sangria frequente, é observada microcitose (diminuição dos glóbulos vermelhos).
Bom efeito terapêutico fornece sangria. Recomenda-se retirar 200-300 ml de sangue semanalmente até que o nível de TEC caia para o valor desejado. Se houver contra-indicações para sangria, restaure percentagem os glóbulos vermelhos podem ser obtidos diluindo o sangue adicionando-lhe uma parte líquida (soluções de alto peso molecular são administradas por via intravenosa).
Deve-se ter em mente que muitas vezes a sangria leva ao desenvolvimento anemia por deficiência de ferro, em que são observados sintomas correspondentes e aumento na contagem de plaquetas.
Para uma doença como a policitemia vera, o tratamento envolve seguir uma determinada dieta. Recomenda-se limitar o consumo de carne e produtos de peixe, por conterem grande quantidade de proteínas, o que estimula ativamente a atividade dos órgãos hematopoiéticos. Você também deve recusar comidas gordurosas. O colesterol contribui para o desenvolvimento da aterosclerose, resultando em coágulos sanguíneos, que já se formam em grandes quantidades em pessoas que sofrem de policitemia.

Além disso, se for diagnosticada policitemia, o tratamento pode incluir quimioterapia. É usado para aumento da trombocitose e coceira intensa. Via de regra, trata-se de um “agente citorredutor” (o medicamento “Hidroxicarbamida”).
Até recentemente, injeções de isótopos radioativos (geralmente fósforo-32) eram usadas para suprimir a medula óssea. Hoje, esse tratamento é cada vez mais recusado devido a alta velocidade transformação leucêmica.
A terapia também inclui injeções de interferon, no tratamento da trombocitose secundária é utilizado o medicamento Anagrelide.
Para esta patologia, muito raramente é realizada, pois a policitemia é uma doença que não é fatal, desde que, claro, tratamento adequado e acompanhamento constante.
A policitemia é uma patologia cujos sintomas podem ser encontrados em recém-nascidos. Esta doença é uma resposta do corpo do bebê à hipóxia, que poderia ter sido provocada.Para corrigir a hipóxia, o corpo do bebê começa a sintetizar um grande número de glóbulos vermelhos.
Além do quadro respiratório, os recém-nascidos também podem desenvolver policitemia vera. Os gêmeos estão especialmente em risco desse risco.
A policitemia no recém-nascido desenvolve-se nas primeiras semanas de vida, suas primeiras manifestações são aumento do hematócrito (até 60%) e aumento significativo dos níveis de hemoglobina.

A policitemia neonatal apresenta vários estágios: inicial, proliferação e depleção. Vamos descrevê-los brevemente.
O estágio inicial da doença praticamente não apresenta manifestações clínicas. A policitemia em uma criança pode ser detectada nesta fase apenas através do exame dos parâmetros do sangue periférico: hematócrito, hemoglobina e nível de glóbulos vermelhos.
Na fase de proliferação, desenvolve-se um aumento do fígado e do baço. Fenômenos pletóricos são observados: pele adquirir uma tonalidade característica “vermelho pletórico”, a criança demonstra ansiedade ao tocar a pele. A síndrome pletórica é complementada por trombose. Nos exames são observadas alterações no número de hemácias, plaquetas e alterações de leucócitos. A contagem de todas as células sanguíneas também pode aumentar, um fenômeno chamado “panmielose”.
A fase de exaustão é caracterizada por perda significativa de peso corporal, astenia e exaustão.
Para um recém-nascido, tais alterações clínicas são extremamente graves e podem provocar mudanças irreversíveis e morte subsequente. A policitemia pode causar uma interrupção na produção de um certo tipo de glóbulo branco, responsável por sistema imunológico corpo. Como resultado, o bebê desenvolve infecções bacterianas graves, que levam à morte.
Depois de ler este artigo, você aprendeu mais sobre uma patologia como a policitemia. Revisamos os sintomas e o tratamento com o máximo de detalhes possível. Esperamos que você considere as informações fornecidas úteis. Cuide-se e seja saudável!
Hepatoesplenomegalia também pode se desenvolver. O diagnóstico é estabelecido com base em um exame de sangue geral, testando a presença de mutações no gene 1AK2 e critérios clínicos. O tratamento inclui o uso de aspirina em baixas doses em todos os pacientes e medicamentos mielossupressores em pacientes de alto risco. A sangria costumava ser o tratamento padrão, mas seu papel agora é controverso.
A policitemia vera é o distúrbio mieloproliferativo mais comum. Sua incidência nos Estados Unidos é de 1,9/100.000, e o risco aumenta com a idade. IP ocorre com um pouco mais de frequência em homens. IP é muito raro em crianças.
Na IP, há aumento da proliferação de todas as linhagens celulares. A este respeito, a PV é por vezes chamada de panmielose devido ao aumento de representantes de todas as 3 linhagens de células do sangue periférico. O aumento da produção de uma linhagem de eritrócitos é chamado de eritrocitose. A trombocitose isolada pode ser observada com PV, mas ocorre mais frequentemente por outros motivos (eritrocitose secundária).
A hematopoiese extramedular pode ocorrer no baço, fígado e outros órgãos que podem servir como local para a formação de células sanguíneas. A renovação das células do sangue periférico aumenta. Em última análise, a doença pode entrar numa fase debilitante, cujas manifestações são indistinguíveis da mielofibrose primária. A transformação para leucemia aguda é rara, mas o risco aumenta com o uso de agentes alquilantes e fósforo radioativo. Este último deve ser usado apenas em casos raros ou nunca.
Complicações. Com IP, o volume de sangue circulante aumenta e sua viscosidade aumenta. Os pacientes são propensos a desenvolver trombose. A trombose pode ocorrer na maioria dos vasos, causando acidentes vasculares cerebrais, ataques isquêmicos transitórios ou síndrome de Budd-Chiari. No passado, os especialistas acreditavam que o aumento da viscosidade do sangue era um fator de risco para trombose. Estudos recentes indicam que o risco de trombose pode depender principalmente da gravidade da leucocitose. No entanto, esta hipótese ainda precisa ser testada em estudos prospectivos desenhados especificamente para esse fim.
A função plaquetária pode ser prejudicada, aumentando o risco de sangramento. O aumento da renovação celular pode causar aumento dos níveis de ácido úrico, aumentando assim o risco de gota e pedras nos rins.
Fatores genéticos. A hematopoiese clonal é característica distintiva IP. Isto indica que a causa da proliferação é uma mutação nas células-tronco hematopoiéticas. A mutação JAK2 V617F (ou uma das várias outras mutações mais raras do gene JAK2) é encontrada em quase todos os pacientes com PV. No entanto, pode-se dizer com quase total certeza que existem outras mutações subjacentes à doença. Eles mantêm a proteína JAK2 num estado de atividade constante, o que leva à proliferação celular excessiva, independentemente da concentração de eritropoietina.
É detectado por acaso pela hemoglobina elevada ou por sintomas de aumento da viscosidade, como fadiga, perda de concentração, dores de cabeça, tonturas, visão escura, coceira na pele, sangramento nasal. Às vezes, manifesta-se como doenças das artérias periféricas ou danos nos vasos sanguíneos do cérebro. Os pacientes costumam ser pletóricos e a maioria apresenta baço aumentado palpável. Podem ocorrer trombose e frequentemente úlceras pépticas, às vezes complicadas por sangramento.
A policitemia vera costuma ser assintomática. Às vezes, um aumento no número de glóbulos vermelhos circulantes e um aumento na viscosidade são acompanhados de fraqueza, tontura, visão turva, fadiga e falta de ar. Um sintoma comum há coceira, especialmente depois do banho. Pode haver rubor facial e veias retinianas dilatadas, bem como vermelhidão e sensibilidade nas palmas das mãos e plantas dos pés, às vezes acompanhada de isquemia digital (eritromelalgia). Hepatomegalia é frequentemente observada; esplenomegalia (às vezes pronunciada) ocorre em 75% dos pacientes.
A trombose pode causar sintomas na área afetada (por exemplo, anormalidade neurológica no acidente vascular cerebral ou ataque isquêmico transitório, dor nas pernas, inchaço das pernas ou ambos na trombose vascular das extremidades inferiores, perda unilateral da visão na trombose vascular retiniana).
O sangramento ocorre em 10% dos pacientes.
O metabolismo acelerado pode causar febre baixa e levar à perda de peso, o que indica a transição da doença para a fase de emaciação. Esta última é clinicamente indistinguível da mielofibrose primária.
A suspeita de PV muitas vezes surge já na fase de um hemograma completo, mas também deve surgir na presença de sintomas correspondentes, em particular a síndrome de Budd-Chiari (vale a pena notar, no entanto, que em alguns pacientes a síndrome de Budd-Chiari se desenvolve antes que o hematócrito aumente). Leucocitose neutrofílica e trombocitose são manifestações comuns, mas não obrigatórias. Pacientes com aumento isolado de hemoglobina ou eritrocitose também podem ter PV, mas nesses casos a eritrocitose secundária deve ser descartada primeiro. PV também pode ser suspeitada em alguns pacientes com níveis normais de hemoglobina, mas com microcitose e sinais de deficiência de ferro. Esta combinação de características pode ocorrer quando a hematopoiese ocorre na presença de reservas limitadas de ferro, o que é uma marca registrada de alguns casos de PV.
A OMS desenvolveu novos critérios de diagnóstico. Portanto, os pacientes com suspeita de PV geralmente devem ser testados para mutações no gene JAK2.
Testar uma amostra de medula óssea nem sempre é necessário.
Nos casos em que é realizada, a panmielose, o grande tamanho e o apinhamento de megacariócitos costumam chamar a atenção na medula óssea. Em alguns casos, são encontradas fibras de reticulina. No entanto, nenhuma alteração na medula óssea permite distinguir IP de outros com absoluta certeza. condições patológicas(por exemplo, policitemia familiar congênita), acompanhada de eritrocitose.
As concentrações plasmáticas de eritropoetina em pacientes com PV são geralmente baixas ou limite inferior normas. Uma concentração aumentada indica a natureza secundária da eritrocitose.
Em alguns casos, a formação de colônias endógenas in vitro de células eritróides (precursores de glóbulos vermelhos retirados do sangue periférico ou da medula óssea de pacientes com PV, em oposição aos de pessoas saudáveis pode formar células eritróides em cultura sem adição de eritropoietina).
A determinação da massa total de eritrócitos usando eritrócitos marcados com cromo pode ajudar a distinguir a policitemia vera da policitemia relativa e pode ajudar a diferenciar a policitemia dos distúrbios mieloproliferativos. Contudo, a técnica para realizar este teste é complexa. Não é realizado rotineiramente devido à sua disponibilidade limitada e ao fato de ser padronizado para uso apenas ao nível do mar.
Para desvios inespecíficos parâmetros laboratoriais que podem ser observados na PV incluem aumento na concentração de vitamina B 12 e aumento na capacidade de ligação de B 12, bem como hiperuricemia e hiperuricosúria (presente em > 30% dos pacientes), aumento da expressão do gene PRV-1 em leucócitos, diminuição da expressão do gene C-mpl (receptor de trombopoietina) em megacariócitos e plaquetas. Esses testes não são necessários para fazer um diagnóstico.
O diagnóstico de policitemia é discutido na subseção " Maior conteúdo hemoglobina." Para o diagnóstico, é importante um aumento da massa eritrocitária na ausência de causas de eritrocitose secundária e esplenomegalia. As contagens de neutrófilos e plaquetas estão frequentemente aumentadas, um cariótipo anormal pode ser detectado na medula óssea e a cultura in vitro da medula óssea exibe crescimento autônomo na ausência de suplementação de fator de crescimento.
Em geral, a PV está associada a uma menor esperança de vida. A sobrevida média para todos os pacientes é de 8 a 15 anos, embora muitos vivam muito mais. Razão comum a morte é trombose. As próximas complicações mais comuns são a mielofibrose e o desenvolvimento de leucemia.
A sobrevida média após o diagnóstico para pacientes em tratamento excede 10 anos. Alguns pacientes vivem mais de 20 anos; entretanto, complicações cerebrovasculares e coronarianas ocorrem em 60% dos pacientes. A doença pode progredir para outro distúrbio mieloproliferativo; a mielofibrose se desenvolve em 15% dos pacientes. A leucemia aguda aparece principalmente em pacientes tratados com fósforo radioativo.
A terapia deve ser individualizada, levando em consideração idade, sexo, estado de saúde, manifestações clínicas e resultados de estudos hematológicos. Os pacientes são divididos em grupo de alto risco e grupo de baixo risco. O grupo de alto risco inclui pacientes >60 anos de idade com história de trombose ou ataque isquêmico transitório, ou ambos.
Aspirina. A aspirina reduz o risco de trombose. Portanto, pacientes submetidos a flebotomia ou apenas flebotomia devem receber aspirina. Mais altas doses aspirina está associada a inaceitáveis alto risco sangramento.
Sangramento. A flebotomia foi a base do tratamento para pacientes de grupos de alto e baixo risco porque os especialistas acreditavam que reduzia a probabilidade de trombose. A justificativa para a flebotomia é atualmente controversa, pois novas pesquisas indicam que os níveis de hemoglobina podem não estar correlacionados com o risco de trombose. Alguns médicos não seguem mais diretrizes rígidas em relação à flebotomia. A flebotomia ainda é uma das alternativas possíveis para qualquer paciente. Numa pequena proporção de pacientes com rubor na pele e aumento da viscosidade sanguínea, a flebotomia pode reduzir os sintomas. O limiar padrão de hematócrito acima do qual a flebotomia é realizada é >45% em homens e >42% em mulheres. Quando o valor do hematócrito cai abaixo do limite, ele é verificado mensalmente e mantido no mesmo nível por flebotomias adicionais, que são realizadas conforme necessário. Se necessário, o volume intravascular é reabastecido com soluções cristalóides ou coloides.
A terapia mielossupressora é indicada para pacientes de alto risco.
O fósforo radioativo (32P) tem sido usado há muito tempo para tratar PV. A eficácia do tratamento varia de 80 a 90%. O fósforo radioativo é bem tolerado e requer menos visitas ao consultório quando o controle da doença é alcançado. No entanto, o uso de fósforo radioativo está associado a um risco aumentado de desenvolvimento de leucemia aguda. A leucemia que ocorre após essa terapia é frequentemente resistente à terapia de indução e é sempre incurável. Assim, o uso do radiofósforo requer seleção cuidadosa dos pacientes (por exemplo, o medicamento deve ser prescrito apenas para aqueles pacientes cuja expectativa de vida devido a patologia concomitante não exceda 5 anos). Deve ser prescrito apenas em casos raros. Muitos médicos nem usam isso.
A hidroxiureia inibe a enzima ribonucleosídeo difosfato redutase. Também é usado para suprimir a atividade da medula óssea. Não existem dados claros sobre a capacidade da hidroxiureia em provocar leucemia. Porém, a possibilidade de transformação em leucemia existe, embora seja pequena. Os pacientes são submetidos a exames de sangue semanais. Depois de atingir um estado estacionário, os intervalos entre os exames de sangue aumentam para 2 semanas e depois para 4 semanas. Se a sua contagem de glóbulos brancos cair<4000/мкл или уровень тромбоцитов падает <100 000/мкл, лечение приостанавливают, а когда упомянутые показатели приходят в норму, возобновляют в дозе на 50% меньше исходной. Дозу гидроксимочевины рационально титровать до достижения практически нормальной величины гематокрита, однако данные в пользу такого титрования отсутствуют. Нормализация уровня лейкоцитов, вероятно, более важна, но как и в предыдущем случае, эта гипотеза не была подтверждена проспективными исследованиями. Подтверждения тому, что нормализация уровня тромбоцитов необходима, нет, и некоторые врачи не увеличивают дозу гидроксимочевины до тех пор, пока число тромбоцитов остается <1,5 млн/мкл. Острая токсичность - нередкое явление. В некоторых случаях у пациентов возникает сыпь, лихорадка, изменения внешнего вида ногтей, кожные язвы.
O interferon alfa-2b é utilizado nos casos em que a hidroxiureia não consegue manter o nível necessário de células sanguíneas ou quando este é ineficaz. Vale ressaltar que o interferon alfa-2b peguilado é geralmente bem tolerado. Este medicamento tem como alvo a doença em nível molecular e tem toxicidade relativamente baixa.
Os medicamentos alquilantes podem provocar o desenvolvimento de leucemia, por isso devem ser evitados.
Vários inibidores da via JAK2 estão atualmente em desenvolvimento clínico. Eles são estudados principalmente em pacientes com mielofibrose em estágios avançados.
Tratamento de complicações. A hiperuricemia é corrigida com alopurinol se concentrações elevadas de ácido úrico forem acompanhadas de sintomas ou se os pacientes estiverem recebendo terapia mielossupressora concomitante. Você pode tentar controlar a coceira com anti-histamínicos, mas às vezes isso pode ser difícil de conseguir. A mielossupressão costuma ser o método mais eficaz. Exemplos de terapia potencialmente eficaz incluem colestiramina, ciproheptadina, cimetedina ou paroxetina.
A sangria alivia rapidamente os sintomas de hiperviscosidade. São retirados 400-500 ml de sangue - e a venesecção é repetida a cada 5-7 dias até que o hematócrito diminua 45%, retirando 400-500 ml de sangue a cada procedimento (menos se o paciente for idoso). A sangria menos frequente, mas regular, mantém esse nível até que a hemoglobina diminua devido à deficiência de ferro. A mieloproliferação subjacente é suprimida com hidroxicarbamida ou interferon. O tratamento com fósforo radioativo (5 mCi 32P por via intravenosa) é reservado para pacientes mais idosos, pois aumenta em 6 a 10 vezes o risco de transformação em leucemia aguda. O tratamento da proliferação da medula óssea pode reduzir o risco de oclusão vascular, controlar o tamanho do baço e reduzir a transformação em mielofibrose. A aspirina reduz o risco de trombose.
– hemoblastose crônica, que se baseia na proliferação ilimitada de toda mielopoiese, predominantemente eritrocitária. Clinicamente, a policitemia se manifesta por sintomas cerebrais (peso na cabeça, tontura, zumbido), síndrome trombohemorrágica (trombose arterial e venosa, sangramento), distúrbios microcirculatórios (frieza dos membros, eritromelalgia, hiperemia da pele e mucosas). As informações diagnósticas básicas são obtidas a partir do estudo do sangue periférico e da medula óssea. Para tratar a policitemia, são utilizadas sangrias, eritrocitaférese e quimioterapia.
O desenvolvimento da policitemia é precedido por alterações mutacionais nas células-tronco hematopoiéticas pluripotentes, que dão origem às três linhagens celulares da medula óssea. A mutação mais comum detectada é o gene da tirosina quinase JAK2 com a substituição da valina pela fenilalanina na posição 617. Às vezes há incidência familiar de eritremia, por exemplo, entre judeus, o que pode indicar uma correlação genética.
Na policitemia, existem 2 tipos de células precursoras hematopoiéticas eritróides na medula óssea: algumas delas se comportam de forma autônoma, sua proliferação não é regulada pela eritropoietina; outros, como esperado, são dependentes de eritropoietina. Acredita-se que a população autônoma de células nada mais é do que um clone mutante - principal substrato da policitemia.
Na patogênese da eritremia, o papel principal pertence ao aumento da eritropoiese, que resulta em eritrocitose absoluta, comprometimento das propriedades reológicas e de coagulação do sangue, metaplasia mieloide do baço e do fígado. A alta viscosidade do sangue causa tendência à trombose vascular e danos hipóxicos aos tecidos, e a hipervolemia causa aumento do suprimento de sangue aos órgãos internos. No final da policitemia, observa-se depleção da hematopoiese e mielofibrose.
Na hematologia, existem 2 formas de policitemia - verdadeira e relativa. A policitemia relativa se desenvolve com contagens normais de glóbulos vermelhos e diminuição do volume plasmático. Essa condição é chamada de estresse ou falsa policitemia e não é discutida no escopo deste artigo.
A policitemia vera (eritremia) pode ser de origem primária ou secundária. A forma primária é uma doença mieloproliferativa independente, baseada em danos à linhagem mieloide da hematopoiese. A policitemia secundária geralmente se desenvolve com atividade aumentada da eritropoetina; esta condição é uma reação compensatória à hipóxia geral e pode ocorrer com patologia pulmonar crônica, defeitos cardíacos “azuis”, tumores adrenais, hemoglobinopatias, ao subir em altitude ou fumar, etc.
A policitemia vera passa por 3 estágios de desenvolvimento: inicial, avançado e terminal.
Estágio I(inicial, assintomático) – dura cerca de 5 anos; é assintomático ou com manifestações clínicas minimamente expressas. Caracterizado por hipervolemia moderada, eritrocitose leve; O tamanho do baço é normal.
Estágio II(eritrêmico, expandido) é dividido em dois subestágios:
Estágio III(anêmico, posteritrêmico, terminal). Caracterizada por anemia, trombocitopenia, leucopenia, transformação mieloide do fígado e baço, mielofibrose secundária. Possíveis resultados da policitemia em outras hemoblastoses.
A eritremia se desenvolve gradualmente durante um longo período de tempo e pode ser detectada acidentalmente durante um exame de sangue. Os primeiros sintomas, como peso na cabeça, zumbido, tontura, visão turva, membros frios, distúrbios do sono, etc., são frequentemente atribuídos à idade avançada ou a doenças concomitantes.
A característica mais característica da policitemia é o desenvolvimento da síndrome pletórica, causada por pancitose e aumento do volume sanguíneo. A evidência de pletora é telangiectasia, coloração vermelho-cereja da pele (especialmente rosto, pescoço, mãos e outras áreas abertas) e membranas mucosas (lábios, língua), hiperemia da esclera. Um sinal diagnóstico típico é o sinal de Cooperman - a cor do palato duro permanece normal, mas o palato mole adquire uma tonalidade cianótica estagnada.
Outro sintoma característico da policitemia é a coceira na pele, que se intensifica após procedimentos hídricos e às vezes se torna insuportável. As manifestações específicas da policitemia também incluem eritromelalgia - uma sensação dolorosa de queimação nas pontas dos dedos, acompanhada de hiperemia.
No estágio avançado da eritremia podem ocorrer enxaquecas dolorosas, dores ósseas, cardialgia e hipertensão arterial. 80% dos pacientes apresentam esplenomegalia moderada ou grave; o fígado aumenta com menos frequência. Muitos pacientes com policitemia notam aumento do sangramento nas gengivas, hematomas na pele e sangramento prolongado após a extração dentária.
A consequência da eritropoiese ineficaz na policitemia é um aumento na síntese de ácido úrico e uma violação do metabolismo das purinas. Isto encontra expressão clínica no desenvolvimento da chamada diátese de urato - gota, urolitíase, cólica renal.
O resultado da microtrombose e da violação do trofismo da pele e das membranas mucosas são úlceras tróficas de perna, úlceras gástricas e duodenais. As complicações mais comuns na clínica de policitemia são trombose vascular de veias profundas, vasos mesentéricos, veias portais, artérias cerebrais e coronárias. Complicações trombóticas (EP, acidente vascular cerebral isquêmico, infarto do miocárdio) são as principais causas de morte em pacientes com policitemia. Ao mesmo tempo, juntamente com a formação de trombos, os pacientes com policitemia são propensos à síndrome hemorrágica com desenvolvimento de sangramento espontâneo em vários locais (gengival, nasal, das veias esofágicas, gastrointestinal, etc.).
As alterações hematológicas que caracterizam a policitemia são decisivas no diagnóstico. Um exame de sangue revela eritrocitose (até 6,5-7,5x10 12 /l), aumento da hemoglobina (até 180-240 g/l), leucocitose (acima de 12x10 9 /l), trombocitose (acima de 400x10 9 /l). A morfologia dos eritrócitos, via de regra, não se altera; com aumento do sangramento, a microcitose pode ser detectada. A confirmação confiável da eritremia é um aumento na massa de glóbulos vermelhos circulantes superior a 32-36 ml/kg. Neurologista, cardiologista, gastroenterologista, urologista.
Para normalizar o volume do Cco e reduzir o risco de complicações trombóticas, a primeira medida é a sangria. As exfusões de sangue são realizadas no volume de 200-500 ml 2 a 3 vezes por semana, seguidas de reposição do volume sanguíneo retirado com solução salina ou reopoliglucina. A consequência da sangria frequente pode ser o desenvolvimento de anemia por deficiência de ferro. A sangria para policitemia pode ser substituída com sucesso pela eritrocitoférese, que permite que apenas a massa de glóbulos vermelhos seja removida da corrente sanguínea, devolvendo o plasma.
Em caso de alterações clínicas e hematológicas pronunciadas, desenvolvimento de complicações vasculares e viscerais, recorrem à terapia mielossupressora com citostáticos (busulfan, mitobronitol, ciclofosfamida, etc.). Às vezes, é administrada terapia com fósforo radioativo. Para normalizar o estado de agregação do sangue, são prescritos heparina, ácido acetilsalicílico, dipiridamol sob o controle de um coagulograma; para hemorragias, estão indicadas transfusões de plaquetas; para diátese de urato - alopurinol.
O curso da eritremia é progressivo; a doença não é propensa a remissões espontâneas e cura espontânea. Os pacientes são forçados a ficar sob a supervisão de um hematologista por toda a vida e a passar por cursos de terapia de hemoexfusão. Na policitemia existe um alto risco de complicações tromboembólicas e hemorrágicas. A incidência de transformação da policitemia em leucemia é de 1% em pacientes que não foram submetidos a tratamento quimioterápico e de 11 a 15% naqueles que recebem terapia citotóxica.

Quantas vezes vemos esse monstro com chifres de cabra quando distribuímos cartas de Tarô. "Diabo" é a personificação do inferno e da morte...

Navegação: Descrição e história do mapa: À beira da estrada existe um grande carvalho, na sua folhagem os pássaros encontram abrigo e alimento...

Caros visitantes do fórum, estou abrindo este tópico para os fãs do baralho "Deviant Moon Tarot" de Patrick Walesa. Tarot...
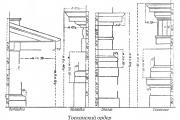
Ordem toscana. Ordem Toscana, uma das cinco ordens arquitetônicas romanas. O nome está associado à arquitetura etrusca...

LIBRETO. Se você ama música (o que sem dúvida ama, já que precisava deste livro), então provavelmente já teve...

Religião: paganismo Nascimento: 942 (0942) Morte: Marcha no Dnieper Família: Rurikovich Pai: Igor...

Masaccio (na verdade Tommaso di Giovanni di Simone Cassai (Guidi), Tommaso di ser Giovanni di Guidi; 21 de dezembro...

(Publicado em Tel Aviv em 1958. Transmitido com pequenas abreviaturas). Judeus! Amem-se uns aos outros,...

Pegue uma concha ou panela e despeje leite e creme nela. Adicione o açúcar baunilhado (açúcar com sementes de baunilha). EM...

Olá pessoal! Hoje temos diante de nós uma receita muito saborosa e simples - salsichas em massa no forno. Você sente...

O abacate é uma das frutas mais saudáveis. Este é um verdadeiro depósito de vitaminas e nutrientes. E ele...

O porco assado é um prato ideal, saboroso e satisfatório, que pode alimentar toda a família. Senhoras...

Este bolo é muito fácil de fazer e tem gosto de clássico Napoleão. Na hora de preparar o bolo existe...

Os representantes desses signos do zodíaco nem sempre se tornam pessoas próximas, mas são agradáveis e interessantes uns com os outros....

Navegação: Descrição e história do mapa: À beira da estrada existe um grande carvalho, na sua folhagem encontram abrigo e alimento...

Caros visitantes do fórum, estou abrindo este tópico para os fãs do baralho "Deviant Tarot" de Patrick Walesa...