Философско виждане на проблема
Смисълът на живота е свързан с въпроса „За какво да живеем“, а не с въпроса как да поддържаме живота. Отношението на човек е...
Едно от редките и изключително опасни заболявания за човешкото здраве и живот е латералната амиотрофична склероза. Това е патология на нервната система, в която двигателните неврони гръбначен мозък, както и кората и мозъчния ствол, претърпяват необратими промени. Заболяването е хронично и се характеризира с постоянна прогресия. Патологичният процес не е много разпространен: на 100 000 души се регистрират приблизително 4-6 случая на заболяването.
Амиотрофичната латерална склероза (ALS) е нарушение във функционирането на нервната система, което провокира постоянно прогресиращо мускулна слабост, което възниква при условия на селективно увреждане на двигателните неврони. Патологията има няколко други имена, например болестта на Лу Гериг, кръстена на световния шампион по бейзбол, който страда от това заболяване. Друго име за страна амиотрофична склероза- Болест на Шарко. Свързва се с името на френския психиатър Жан-Мартен Шарко, описал патологичната мускулна слабост през втората половина на 19 век.
И накрая, ALS е известно като заболяване на моторните неврони според ICD-10.

Понякога, когато описвам това патологично състояние, думата „странично” се заменя с епитета „странично”. Включването на това прилагателно в името на заболяването се дължи на факта, че невроните, разположени в страничните проекции на гръбначния мозък, са най-податливи на промени.
Патологичните промени, свързани с отслабване и атрофия на мускулите, са причинени от разрушаването на неврони, които са отговорни за предаването на сигнали от мозъка към мускулите. При това заболяване могат да бъдат засегнати както централните, така и периферните моторни неврони. Първият от тях се намира в кората мозъчни полукълбамозък Когато е повреден, се развива мускулна слабост, докато тонусът се повишава и рефлексите се засилват. Периферният двигателен неврон е разположен в мозъчния ствол и на различни нивагръбначен мозък. Ако претърпи патологични промени, тогава се наблюдава развитие на мускулна слабост, но в същото време намаляват рефлексите, както и мускулния тонус, както и развитието на мускулна атрофия.
При условия на увреждане на един от моторните неврони (или и двата наведнъж) предаването на импулси от него към мускула е блокирано.
Амиотрофичната латерална склероза се проявява в нарастваща мускулна слабост и мускулна загуба, което в резултат на хроничен ход, води до пълно обездвижване на пациента и нарушени дихателни функции.

В момента в света има около 70 000 души, диагностицирани с ALS. Обикновено тази патология се проявява след 40-50 години. Според клиничните данни обикновено води до смърт в рамките на 5 години след диагностицирането. Известният физик Стивън Хокинг обаче живее с това заболяване вече 50 години.
Само 7% от всички пациенти живеят повече от 60 месеца.
Установено е, че мъжете страдат от болестта на Шарко по-често от жените. Наскоро учени предположиха, че най-голямото числослучаи на патология се регистрират при хора с висок интелект и спортисти, които не са имали сериозни здравословни проблеми през целия си живот.
Амиотрофичната латерална склероза стана известна на медицината не много отдавна и основната причина за това заболяване все още не е ясна. Изследователите обаче са съгласни, че се основава на натрупването на патологичен неразтворим протеин в двигателните клетки на нервната система. Това е, което води до тяхната смърт.
Има теория, според която водещата роля на развитието необратими променипринадлежи към промяна в свойствата на специален ензим, чиято функция е да предпазва клетките на тялото от разрушаване от кислородни радикали. Този ензим се нарича супероксид дисмутаза-1. Такива промени са свързани с генни мутации, които в 25% от случаите са наследени. Именно този факт мотивира хипотезата за генетична природаБАС.
Възможно е разрушаването на моторните неврони да е свързано с мутации в други структури, например образувания, които придават формата на нервната клетка и осигуряват нейната рамка.
Когато описват природата на амиотрофичната латерална склероза, те споменават следните причининеговото развитие:
Водещи на предаването „Живей здравословно! ще говорим за неизлечима болестповече информация:
Лекарите включват следните рискови фактори:
Амиотрофичната латерална склероза не е заразна болест, не могат да се заразят от друго лице.
Заболяването на моторните неврони се класифицира в типове въз основа на тежестта на увреждането на нервните клетки. Проявите на всеки един от тях са в основата си сходни, но докато се развиват патологичен процес, разликата става по-очевидна.
В неврологията има следните формизаболявания:

Снимката показва прогресивна булбарна парализа
Прогресията на амиотрофичната латерална склероза причинява усложнения, които водят до смъртта на пациента. Те включват:
Заболяването започва с увреждане на крайниците, което след това се разпространява в останалата част от тялото.
Ранните симптоми на амиотрофична латерална склероза са:
Уважаеми читатели, за основните причини и симптоми на заболяването гледайте видеоклипа по-долу:
С напредването на амиотрофичната латерална склероза пациентът забелязва скованост на мускулите, което е свързано с повишаване на техния тонус и трудност при опитите им да се отпуснат. Има и такива характерни особености, като дисбаланс, спонтанни пристъпи на смях или плач, ограничени движения на езика, промяна в гласа.
В по-късните стадии на заболяването се наблюдават прекъсвания на дишането, депресия и невъзможност за самостоятелно движение.
Диагнозата на амиотрофичната латерална склероза изисква голям брой специфични мерки. Първо, пациентът се преглежда и интервюира от невролог, който се интересува от следната информация:
Други методи, които ви позволяват да направите правилна диагноза, включват:

Процедурата за извършване на лумбална пункция включва пробиване на арахноидната мембрана на гръбначния мозък между 3-ти и 4-ти или 2-ри и 3-ти лумбален прешлен с игла на Beer за събиране на цереброспинална течност.
В допълнение към изброените методи трябва да се направи диференциална диагноза. Амиотрофичната латерална склероза трябва да се разграничава от патологии като тумор на гръбначния мозък, цервикална миелопатия, синдром на малабсорбция и диабетна амиотрофия.
Лечението на амиотрофичната латерална склероза има за цел да забави прогресията на патологичния процес, тъй като настъпилите промени са необратими. Заболяването не може да се излекува.
Основното и единствено лекарство, което в момента се препоръчва за забавяне на прогресията на заболяването, е Riluzole или Rilutek. Основната му активна съставка предотвратява по-нататъшното увреждане на невроните, като по този начин забавя прогресията на заболяването.
Други лекарства се използват единствено за облекчаване на преобладаващите симптоми, които влошават качеството на живот на пациента. Те включват:

Различни видове действие на мускулните релаксанти
Пациент с диагноза ALS се нуждае от устройства, които му позволяват да се движи (инвалидна количка, проходилка, легло с различни функции, по-специално оборудван с асансьор). За да може пациентът да диша, може да се извърши специална операция - трахеостомия. По време на манипулация в трахеята хирургичносъздайте дупка.
Пациентът трябва да бъде осигурен пълна грижа. Особено внимание трябва да се обърне на мерките за предотвратяване на образуването на рани от залежаване. Леглото винаги трябва да е сухо и чисто, както и тялото на пациента.
Друг неразделен компонент на поддържащата терапия за амиотрофична латерална склероза е работата с психолог. Помощта на специалист ще е необходима не само на пациента, но и на членовете на неговото семейство.

Поради увреждане, дължащо се на ALS, пациентът трябва да използва инвалидна количка
Прогнозата за това специфично заболяване е лоша. Болестта непрекъснато прогресира, като все повече влошава качеството на живот на пациента и в крайна сметка води до остра дихателна недостатъчност и смърт на пациента. В зависимост от формата, в която протича патологичният процес, пациентът може да живее от 2 до 12 години. При булбарната форма, както и ако пациентът е в напреднала възраст, продължителността на живота се намалява до 1-3 години.
Амиотрофичната латерална склероза е рядко заболяване, което непрекъснато прогресира и неизбежно води до смърт на пациента. Причините за това явление не са напълно проучени, има само предположения относно вероятните рискови фактори. Лечението на ALS се свежда до облекчаване на състоянието на пациента и осигуряване на най-удобните условия на живот.
Амиотрофична латерална склероза (ALS), наричана още моторна невронна болест или болест на Шарко-Кожевников, моторна невронна болест и на някои места по света болестта на Лу Гериг, която засяга главно езикоговорящите региони. английски език. Уважаеми пациенти, в тази връзка не бива да се учудвате или съмнявате, ако в текста на нашата статия срещнете различни наименования на този много лош патологичен процес, което води първо до пълна инвалидност, а след това и до смърт.
В основата на това ужасно заболяване са лезии на мозъчния ствол, които не спират в тази област, а се разпространяват към предните рога на гръбначния мозък (нивото на цервикалното удебеляване) и пирамидните пътища, което води до дегенерация на скелетната мускулатура. В хистологичните препарати се откриват цитоплазмени включвания, наречени тела на Бунин, а на фона на съдови инфилтрати се наблюдават дегенеративни промени, набръчкани и мъртви нервни клетки, на мястото на които растат глиални елементи. Очевидно е, че процесът, в допълнение към всички части на главния и гръбначния мозък (малък мозък, мозъчен ствол, кора, подкортекс и др.), Моторните ядра на черепните нерви (черепните нерви) засяга менинги, мозъчните съдове и гръбначното съдово легло. По време на аутопсията патологът отбелязва, че цервикалното и лумбалното удебеляване при пациентите е значително намалено по обем, а багажникът е напълно атрофиран.

Ако преди 20 години пациентите са живели едва 4 години, то в наше време има нарастваща тенденция средна продължителностживот, който вече достига 5-7 години. Церебралната форма все още няма дълголетие (3-4 години), а булбарната форма не дава много шансове (5-6 години). Вярно е, че някои живеят по 12 години, но основно това са пациенти с цервико-торакална форма. Но какво означава този период, ако болестта на Шарко (спорадичните форми) не щади децата (гимназията) и юношеството, докато мъжкият пол има по-голям "шанс" да се сдобие с болест на моторните неврони. Фамилните случаи се появяват по-често в зряла възраст. Реалната опасност от заболяване остава между 40 и 60 години, но след 55 мъжете вече не държат преднина и боледуват като жените.
Булбарните нарушения в дейността на центровете, отговорни за дихателната функция и работата, обикновено водят до смърт. на сърдечно-съдовата система.
В литературата можете да намерите такова определение като „синдром на ALS“. Този синдром няма нищо общо с болестта на моторните неврони, причинява се от напълно различни причини и придружава други заболявания (някои протеинемия и т.н.), въпреки че симптомите на синдрома на ALS много напомнят на ранния стадий на болестта на Лу Гериг, когато клиниката все още не се е развила бързо. По същата причина начална фазаАмиотрофичната латерална склероза се диференцира от () или.
 ALS няма граници в болното човешко тяло, тя се движи по-нататък и по този начин засяга цялото тяло на пациента, поради което формите на амиотрофичната латерална склероза се разграничават доста условно, въз основа на началото на патологичния процес и др. ясни знаципоражения. Точно преобладаващсимптоми по време на амиотрофична латерална склероза, а не изолирани засегнати области, ни позволяват да определим неговите форми, които могат да бъдат представени в следната форма:
ALS няма граници в болното човешко тяло, тя се движи по-нататък и по този начин засяга цялото тяло на пациента, поради което формите на амиотрофичната латерална склероза се разграничават доста условно, въз основа на началото на патологичния процес и др. ясни знаципоражения. Точно преобладаващсимптоми по време на амиотрофична латерална склероза, а не изолирани засегнати области, ни позволяват да определим неговите форми, които могат да бъдат представени в следната форма:
Факторите, които могат да отключат този тежък патологичен процес, не са толкова много, но човек може да се сблъска с всеки един от тях всеки ден, независимо от възрастта, пола и географско местоположение, с изключение, разбира се, на наследственото предразположение, което е характерно само за определена част от населението (5-10%).
И така, причините за заболяването на моторните неврони:
Симптомите на амиотрофичната латерална склероза се характеризират предимно с появата на периферни и централна парезаръце, както е показано със следните знаци:

Очевидно е, че включвайки цялото тяло в процеса, болестта на Шарко дава богата и разнообразна симптоматика, която обаче накратко може да бъде представена от синдроми:
Що се отнася до диагностиката, тя разчита предимно на неврологичния статус, и основната инструментален метод ENMG (електроневромиография) е призната за търсене на ALS; други тестове се извършват за изключване на заболявания с подобни симптоми или за изследване на тялото на пациента, по-специално състоянието на дихателната система и опорно-двигателния апарат. По този начин списъкът необходими изследваниявключва:
Терапията за заболяване на моторните неврони е основно насочена към общо укрепване, поддържане на тялото и облекчаване на симптомите.С развитието на патологичния процес се увеличава дихателната недостатъчност, така че за подобряване на дихателната дейност пациентът първо (докато е в инвалидна количка) преминава към NIV устройство (за неинвазивна вентилация на белите дробове), а след това, когато може вече не се справят, към стационарно вентилационно оборудване.

Наистина ефективно средство за защитавсе още не е изобретен за лечение на амиотрофична латерална склероза, обаче, лечението все още е необходимо и на пациента се предписва лекарствена терапия:
Едва ли може да се спори с твърдението, че пациентът с болестта на Шарко се нуждае от специални грижи. Специално е, защото само храненето си заслужава. Какво ще кажете за борбата с раните от залежаване? Ами депресията? Пациентът е критичен към състоянието си, много се тревожи, че всеки ден състоянието му се влошава и в крайна сметка спира (не по собствена воля) да се грижи за себе си, не може да общува с другите и да се наслади на вкусна вечеря.
Такъв пациент се нуждае от:
Предотвратяването на рани от залежаване е много важно. Те са вътре подобни случаине се карайте да чакате дълго, така че леглото трябва да е чисто и сухо, както и тялото на пациента.
Пациентът се храни предимно с течна, лесна за поглъщане храна, богати на протеинии витамини (стига да се запази функцията за преглъщане). Впоследствие пациентът се храни чрез сонда, след което се прибягва до принудителна, но последна мярка - налагане на гастростоми.

Очевидно е, че пациент с амиотрофична латерална склероза страда много: както морално, така и физически. В същото време страдат и хората, които се грижат за него, за които той е близък човек. Съгласете се, много е трудно да погледнете в избледняващи очи, да видите болка и отчаяние и да не можете да помогнете да победите болестта, да я излекувате, да я върнете към живот. скъп човек. Роднините, които се грижат за такъв болен човек, губят сили и често изпадат в отчаяние и депресивно състояние, и затова се нуждаят и от помощта на психотерапевт с предписване на успокоителни и антидепресанти.
Обикновено в описанието на лечението на всяка болест читателите търсят превантивни мерки и начини да се отърват от определено заболяване народни средства. Всъщност препоръчително алтернативна медицина, съдържащ големи количества витамини от група В, пшеничен зародиш и овесени зърна, орехии прополисът може да не навреди на болния, но и няма да го излекува. Освен това не трябва да забравяме, че такива хора често имат проблеми с преглъщането, т.н в случай на болест на Шарко, не трябва да разчитате на традиционната медицина.
Това е това - амиотрофична латерална склероза (и много други имена за нея). Болестта е страшно коварна, непонятна и нелечима. Може би някой ден човек ще успее да укроти тази болест, поне, да се надяваме на най-доброто, защото учени от цял свят работят по този проблем.
Амиотрофичната латерална склероза (ALS) е нелечимо, прогресиращо заболяване на централната нервна система, при което пациентът изпитва увреждане на горните и долните двигателни неврони, което причинява мускулна атрофия и парализа. Честотата на тази патология е около 2-7 случая на 100 хиляди души. Най-често заболяването се диагностицира при пациенти на възраст над 50 години.
Учените все още не са създали единна цялостна класификация на ALS. Има няколко подхода за класифициране на болестта. Например северноамериканският подход включва идентифициране на следните видове ALS: спорадичен, фамилен, спорадичен ендемичен. Класификацията на амиотрофичната латерална склероза включва следните форми на заболяването: булбарна, лумбосакрална, цервико-торакална и първично генерализирана. Има и няколко варианта на заболяването: смесени, пирамидални и сегментно-ядрени.
Към най-често срещаните начални симптомизаболявания включват крампи (болезнени мускулни спазми), летаргия и слабост в дисталните ръце, булбарни нарушения, мускулна атрофия на краката, слабост в раменния пояс. Освен това за различни вариантиЗаболяването се характеризира с различни клинични прояви.
Точните причини за амиотрофична латерална склероза все още се изследват от учените. Въпреки това могат да бъдат посочени няколко фактора, които провокират заболяването. Например, около 5% от заболяванията са с наследствена етиология. Най-малко 20% от случаите са свързани с мутации в гена на супероксид дисмутаза-1. Учените са го доказали важна роляВисоката активност на глутаматергичната система играе роля в началото на заболяването. Факт е, че излишната глутаминова киселина провокира превъзбуждане и внезапна смърт на невроните. Доказан е и молекулярно-генетичният механизъм на патологията. Причинява се от повишаване на нивото на ДНК и РНК в клетките, което в крайна сметка води до нарушаване на протеиновия синтез.
Учените също така идентифицират няколко предразполагащи фактора, които играят важна роля за появата на ALS. На първо място, такива фактори включват възрастта. Факт е, че заболяването обикновено се развива при пациенти на възраст 30-50 години. Струва си да се помни, че само около 5% от пациентите имат наследствена предразположеност към ALS. В по-голямата част от случаите на ALS причината за патологията не може да бъде установена.
Ранният ход на заболяването се характеризира със симптоми като конвулсии, потрепвания, изтръпване на мускулите, затруднен говор и слабост в крайниците. Тъй като подобни симптоми са характерни за много неврологични заболявания, диагностицирането на ALS на ранен етап е трудно. В повечето случаи заболяването може да се диагностицира на етапа на мускулна атрофия.
В зависимост от засегнатото заболяване различни частитяло, разграничете ALS на крайниците и булбарна ALS. В първия случай пациентите изпитват влошаване на гъвкавостта на глезена, неудобство при ходене и започват да се спъват. Булбарният ALS се проявява със затруднен говор (назален шум, затруднено преглъщане). Скоро пациентът се движи трудно или вече не може да се движи самостоятелно. Обикновено заболяването не оказва вредно въздействие върху умствен капацитетпациент, но води до тежка депресия. В повечето случаи от появата на първите симптоми до смъртта минават около три до пет години.
Тъй като ALS е нелечимо заболяване, което бързо съкращава живота на човека, прегледът на пациента трябва да бъде изчерпателен и точен. Изключително важно е да поставите правилна диагнозапациента, за да започне своевременно облекчаване на основните му симптоми, тъй като това може да удължи живота на пациента. Планът за преглед обикновено включва история на живота и болестта, неврологичен и физикален преглед, ЯМР на гръбначен и главен мозък, ЕМГ и лабораторни изследвания.
Диагностиката на заболяването започва с подробно интервю с пациента. А именно, лекарят трябва да изясни дали пациентът се оплаква от мускулни спазми и потрепвания, слабост и скованост, нарушено движение на ръцете, говор, ходене, преглъщане, слюноотделяне, чести задухи, загуба на тегло, умора, задух по време на тренировка. Освен това лекарят трябва да попита дали пациентът е забелязал двойно виждане, загуба на паметта, усещане за пълзене по тялото или проблеми с уринирането. Задължително е пациентът да се разпита за фамилната му история – дали има роднини с хронични двигателни нарушения.
Основната цел на физикалния преглед е да се оцени конституцията на пациента, да се претегли, да се измери височината му и да се изчисли индексът на телесна маса. Неврологичният преглед обикновено включва невропсихологично изследване. При оценка на булбарните функции лекарят обръща внимание на тембъра на гласа, скоростта на речта, фарингеалния рефлекс, наличието на атрофия на езика и парезата на мекото небце. Освен това по време на прегледа се проверява силата на трапецовидните мускули.
Основният инструментален метод за диагностициране на заболяването е ЕМГ с игла. Тази техника ви позволява да идентифицирате признаци на заболяване като остра или хронична денервация. В ранните стадии на заболяването стимулационната ЕМГ е неефективна, тъй като не открива забележими признаци на ALS.
В процеса на диагностициране на заболяването лекарите използват и невроизобразителни методи. ЯМР на гръбначния и главния мозък играе голяма роля в диференциалната диагноза на ALS. По време на ЯМР при 17-67% от пациентите е възможно да се идентифицират симптоми на дегенерация на пирамидните пътища и атрофия на моторната кора на мозъка. Заслужава обаче да се отбележи, че тази техника е неефективна при диагностициране на заболяването при пациенти с булбарен синдром.
По време на диагностицирането на ALS се извършват много лабораторни изследвания. По-специално, лекарите могат да предписват клинични и биохимични изследванияизследване на кръв, цереброспинална течност, серологични изследвания. Въпреки това, единственият ефективен и надежден метод за анализ все още се счита за молекулярно-генетичен анализ. Наличието на мутации в гена за супероксид дисмутаза-1 се счита за подозрение за ALS.
Тъй като симптомите на амиотрофичната латерална склероза са подобни в много отношения на проявите на други неврологични патологии, лекарите трябва да извършват диференциална диагноза. Най-точната диагноза може да се направи с помощта на ЯМР на мозъка и гръбначния стълб. Преди всичко ALS трябва да се разграничи от мускулните заболявания, които включват дистрофична миотония на Rossolimo-Steinert-Kurshman, миозит с клетъчни аномалии и окулофарингеална миодистрофия.
Също така е необходимо да се разграничи ALS от патологиите на гръбначния мозък:
Диференциалната диагноза също е необходима, за да се разграничи заболяването от системни патологии, лезии на нервно-мускулния синапс и мозъчни патологии като множествена системна атрофия, дисциркулаторна енцефалопатия и сирингобулбия.
Основните цели на лечението на амиотрофична латерална склероза се считат за забавяне на прогресията на заболяването, както и премахване на неговите симптоми, които значително влошават качеството на живот на пациента. Трябва да се помни, че ALS е сериозно нелечимо заболяване, което съкращава продължителността на живота на човека. Ето защо лекарят има право да информира пациента за диагнозата само след цялостен и задълбочен преглед.
Лечението на заболяването включва медикаментозна и нелекарствена терапия. Последното предполага мерки за сигурност. Пациентът трябва да ограничи физическата активност, която може да ускори прогресията на ALS. Освен това е много важно да се храните правилно и питателно. Лекарствената терапия е разделена на два вида: патогенетична и палиативна.
Към днешна дата единственото лекарство, което може да забави прогресията на ALS, е рилузол. Доказано е, че приемът му може да удължи живота на пациента средно с три месеца. Това лекарство е показано за пациенти, чиято продължителност на заболяването е по-малко от 5 години. Пациентът трябва да получава 100 mg от лекарството дневно. За да избегнете риска от лекарствено индуциран хепатит, на всеки три месеца е необходимо да се проверяват нивата на AST, ALT и LDH. Тъй като мъжете и пушачите имат по-ниски концентрации на рилузол в кръвта си, те трябва или да ограничат пушенето, или да го спрат напълно. лош навик. Ще трябва да приемате лекарството цял живот.
Учените многократно са се опитвали да използват други лекарства за патогенетична терапия. Подобни експерименти обаче не се оказаха ефективни. Сред тях бяха:
Ефективността от приема му също не е доказана високи дози Cerebrolysin, въпреки факта, че това лекарство е в състояние леко да подобри състоянието на пациентите.
Палиативната терапия е предназначена да елиминира комплекс от симптоми на заболяването и по този начин да подобри качеството на живот на пациента. За премахване на определени симптоми на ALS се използват следните техники:
За подобряване на мускулния метаболизъм на пациент с ALS могат да бъдат предписани следните лекарства: креатин, карнитин, разтвор на левокарнитин, триметилхидразиниев пропионат. На пациентите е показана и мултивитаминова терапия, която включва прием на мултивитамини (невромултивит, милгама) и тиоктова киселина.
При повечето пациенти с ALS заболяването е придружено от сериозни двигателни увреждания, включително ограничена подвижност. Разбира се, това причинява голям дискомфорт на пациента, който постоянно се нуждае от помощ от други хора. Елиминирайте някои двигателни нарушенияТехниките за ортопедична корекция помагат. Лекарят трябва да обясни на пациента, че използването на помощни средства не показва неговото увреждане, а само намалява затрудненията, причинени от заболяването.
Най-животозастрашаващият симптом на заболяването се счита за дихателна недостатъчност. Най-ранните му симптоми ще бъдат сутрешна умора, ярки сънища, сънливост през деня и неудовлетвореност от съня. За откриване на дихателна недостатъчност на ранен етап се извършва полисомнография и спирография. За премахване на апнея са показани лекарства и неинвазивна вентилация. Доказано е, че тези техники могат да удължат живота на пациента с една година. Ако пациентът се нуждае от асистирано дишане за повече от 20 часа, лекарят поставя въпроса за пълен преход към инвазивна вентилация.
Пациентите, които са преминали първичен преглед или повторно диагностициране на заболяването, трябва да останат подложени амбулаторно наблюдение. При поява на нови симптоми те също трябва да получат квалифициран съвет. Пациентите трябва да приемат повечето лекарства редовно. Само витамини и миотропни лекарства се приемат на курсове на етапи.
На всеки три месеца пациентът трябва да се подложи на спирография. Ако приема редовно рилузол, той трябва да проверява LDH, AST и ALT на всеки шест месеца. Ако пациентът има дисфагия, трябва периодично да се измерват нивата на кръвната захар и трофичния статус. Пациентите имат избор на режим на лечение: те могат да останат у дома или да останат в хоспис.
Прогнозата за пациенти с ALS до голяма степен зависи от хода на заболяването. Доказано е, че около 80-90% от пациентите с тежки респираторни усложнения умират в рамките на 3-5 години след появата на първите признаци на заболяването. Останалите 10% от пациентите имат доброкачествено протичане на заболяването. Продължителността на заболяването е значително намалена при наличие на следните фактори: възрастта на пациента е под 45 години, булбарна поява на ALS, бърза прогресия на заболяването.
Амиотрофичната латерална склероза е сериозно неврологично заболяване, което причинява мускулна слабост, увреждане и в крайна сметка смърт. ALS често се нарича болестта на Лу Гериг, на името на известния бейзболен играч, който е диагностициран през 1939 г. В някои страни ALS и заболяването на моторните неврони понякога се използват взаимозаменяемо.
В световен мащаб ALS засяга 1-3 души на 100 000. В по-голямата част от случаите - от 90 до 95 процента от случаите на това заболяване, лекарите не могат да обяснят причината за това заболяване. Само в 5-10 процента от случаите се проследява генетичната детерминация. ALS често започва с мускулни крампи в ръката или крака и затруднен говор. В крайна сметка ALS нарушава контрола на мускулите, отговорни за дихателните движения при преглъщане.
Ранните симптоми на ALS включват:
Заболяването често започва в ръцете, краката или крайниците и след това се разпространява в други части на тялото. С началото на заболяването симптомите започват да прогресират, мускулите стават по-слаби и след това настъпва парализа. В крайна сметка има нарушение на актовете на дъвчене, преглъщане и дишане.
При амиотрофична латерална склероза нервните клетки, които контролират движението (моторни неврони), започват постепенно да умират, което води до постепенно отслабване на мускулите и тяхната атрофия. ALS се наследява в 5-10 процента от случаите. В други случаи ALS изглежда възниква спонтанно.
В момента се проучват няколко възможни причини за ALS, включително:
Основните рискови фактори за ALS включват:
Факторите на околната среда, които увеличават риска от това заболяване, включват:
 С напредването на заболяването пациентите с амиотрофична латерална склероза изпитват следните усложнения.
С напредването на заболяването пациентите с амиотрофична латерална склероза изпитват следните усложнения.
Ако се появят някои от ранните симптоми на нервно-мускулни заболявания, трябва да се свържете с Вашия лекар, който при необходимост ще Ви насочи за консултация с невролог. Но дори навременното посещение при невролог не гарантира, че диагнозата ще бъде поставена незабавно, тъй като проверката на диагнозата отнема известно време. Неврологът ще се интересува от медицинската история и неврологичния статус.
Амиотрофичната латерална склероза е доста трудна за диагностициране в ранните етапи, тъй като симптомите са подобни на други неврологични заболявания. Използват се следните диагностични методи:
Поради факта, че процесите при амиотрофичната латерална склероза не могат да бъдат обърнати, лечението е насочено към забавяне на прогресията на симптомите.
Медикаментозно лечение. Лекарството рилузол (RILUTEK) е първото и единствено одобрено лекарство за забавяне на ALS. Лекарството има инхибиращ ефект върху прогресията на заболяването при някои пациенти, вероятно чрез намаляване на нивото на глутамат, вещество, което е медиатор в нервната система и чието ниво често е повишено при пациенти с ALS. В допълнение, други лекарства могат да бъдат предписани за облекчаване на симптоми като запек. мускулни крампиумора хиперсаливация болка депресия.
Упражняваща терапия. Физическите упражнения под наблюдението на лекар по физиотерапия могат да помогнат за поддържане на мускулната сила и обхвата на движение за повече дълъг периоддейността на сърдечно-съдовата система и подобряване на общото благосъстояние.
С помощта на проходилка или инвалиден столсъщо ви позволява да поддържате определен диапазон от движения.
Психологическа помощ. Помощта на психолог често се изисква поради осъзнаването на пациента за нелечимостта на заболяването. Въпреки че в някои случаи продължителността на живота може да надхвърли 3-5 години и да достигне 10 години.
ALS синдром означава. Заболяването засяга централната нервна система, развива се бавно, но атакува гръбначния и главния мозък, както и ядрата на черепномозъчните нерви. Ако не се лекува, болестта може да увреди моторните неврони, причинявайки парализа и мускулна атрофия. Нека да разгледаме какво представлява миелопатията и какви са нейните симптоми, причини и как се провежда лечението.
Миелопатията е патология на гръбначния мозък, която може да се развие при синдром на ALS. Ако функционирането на гръбначния мозък е нарушено, функциите и реакциите са недостатъчни, човек е изправен пред тежки последици.
Причини, които провокират появата на миелопатия:
Заболяването може да се развие поради един от тези фактори или поради няколко.
Миелопатията има следните симптоми:

Изброените симптоми изискват лечение. Но преди започване на лечението е важно да се диагностицира миелопатия. За поставяне на диагнозата се използва съвременна апаратура и диференциални тестове.
Миелопатията изисква лечение. Заболяване в остър периодлекувани чрез премахване на болката в засегнатата област. Лечението се извършва с помощта на блокада. Кожата около лезията се инжектира с болкоуспокояващи. Благодарение на блокадата мозъкът не получава сигнал за наличието на възпаление в ставите или мускулите. В резултат на това болката се блокира за дълго време.
След това лечението се извършва с помощта на:
Благодарение на тези мерки благосъстоянието на пациента се подобрява.
Много често миелопатията се развива в хронична форма. В този случай, за да се предотврати прогресирането и обострянето на болестта, е важно да се поддържа болестта в състояние на ремисия.
За да може болестта да ви безпокои възможно най-малко, е важно да се извърши нейната профилактика.
Превантивните мерки са насочени към предотвратяване на причините, които причиняват намаляване на функциите на гръбначния мозък. 
Трябва да се храните правилно, да се стегнете, да се опитате да се отървете от наднормено тегло. За да предотвратите всяка болест, укрепвайте имунната си система по всякакъв начин.
За децата, за да предотвратите заболяване, създайте график за работа и почивка и разпределете товара.
Поради мутацията на протеини с развитието на вътреклетъчни агрегати започва да се формира болестта ALS. Обикновено заболяването засяга хора на възраст между 40 и 60 години.
Точните причини за развитието на болестта не са напълно изяснени. Но учените са установили, че болестта се развива поради появата на четириверижна ДНК в човешките клетки. В резултат на това синтезът на протеини се нарушава и симптомите на синдрома започват да се появяват.
За точна диагноза е важно да посетите няколко специалисти.
5 процента от хората получават заболяването от свои роднини поради наследствен фактор.
Причините за синдрома могат да бъдат като инфекция, нараняване или инфекциозни заболявания.
Повечето хора днес имат симптоми на ALS синдром. Във всеки случай заболяването има свои собствени причини и симптоми на развитие. Важно е да наблюдавате здравето си, за да идентифицирате патологията в ранните етапи на проявление и да започнете нейното лечение. 
Симптоми на синдрома:
Болестта ALS е много трудна за диагностициране в ранните етапи. Но опитни специалисти изучават симптомите, помислете различни версиизаболявания и едва след това се поставя диагноза и се предписва лечение.
Ако пациентът все пак е диагностициран с ALS, неговите роднини трябва да се подготвят за трудности.
Пациентът напълно губи способността си да се движи самостоятелно. След това не може да се храни нормално. Освен това понякога има много силно лигавене. Някои пациенти се нуждаят от специално ентерално хранене.
След известно време започват проблеми с дихателната система и се развива дихателна недостатъчност.
Пациентите често се притесняват от главоболие, задушаване и кошмари. 
Като се има предвид местоположението на мускулите, експертите идентифицират следните форми на ALS синдром:
Булбарната форма на заболяването се характеризира със следните нарушения: изтръпване на езика, парализа на небцето, слабост на дъвкателните мускули. Говорът и способността за нормално преглъщане също са нарушени. IN тежки случаипациентът може да започне да се смее или да плаче без причина, да се движи оживено Долна челюст. След известно време се засягат ръцете и краката. Най-често дори след правилно лечение пациентите умират.
При цервико-торакалната форма на заболяването мускулите на ръцете и краката атрофират.
При лумбосакралната форма възниква атрофична пареза на долните крайници. Когато формата е напреднала, ръцете и черепните мускули също са парализирани.
При церебралната форма крайниците са парализирани и са засегнати периферните двигателни неврони. Пациентът се смее и плаче без причина. Понякога започва активно да движи долната си челюст.
За да се постави диагноза, се извършва електромонограма. Проучването показва, че има ритъм на „ограда“ в потенциалите на фастикулация; скоростта на провеждане не се променя. За правилна диагноза е важно да се проследят всички области на гръбначния стълб:

Важно е да не се бърка синдромът с други видове заболявания, които могат да имат подобни симптоми. Затова точната диагноза трябва да бъде поставена от опитни специалисти.
Лечението на ALS има за цел да облекчи симптомите на заболяването. Терапията се провежда с Riluzone. Но това лекарство се предлага само в Европа и САЩ. Медицината не лекува болестта. Но това помага да се удължи и подобри качеството на живот на болен човек.
Лечението на синдрома на ALS се извършва със следните лекарства:
Работата на Рилузон. Кога се предава? нервен импулс, се освобождава глутамат, химичен медиатор в централната нервна система. Rizulon намалява количеството на екскрецията на такова вещество.
След изследване учените установиха, че излишъкът от глутамат уврежда невроните в гръбначния мозък и мозъка. Според тестовете хората, които използват Rizulon, живеят по-дълго от другите пациенти - с три месеца.
Учените са установили също, че антиоксидантите потискат признаците на синдрома. Тези вещества помагат на тялото да предотврати увреждането на свободните радикали. Антиоксидантите се избират от лекаря, като се вземе предвид здравословното състояние на пациента. 
За да улесним живота на хората със синдрома, съпътстващо лечение. Тъй като лечението на заболяването отнема много време, важно е да се лекува не само основното заболяване, но и други симптоми. Според специалистите релаксацията облекчава безпокойството и страха.
За отпускане на мускулите се използват рефлексотерапия, ароматерапия и масаж. Благодарение на такива процедури се нормализира лимфата и кръвообращението и се премахва болката. Чрез извършването на всички процедури специалистите стимулират ендогенните болкоуспокояващи и ендорфини. Но е важно да се извършват дейности с нервна системаиндивидуално. Ето защо, преди да започнете лечението, посетете лекар и преминете всички прегледи.
Синдромът непрекъснато прогресира и ако не се лекува, може да бъде фатален. След като се открият признаци на заболяването, пациентът има възможност да живее още пет години. Но за да протича добре животът му, осигурете поддържаща терапия.
Неблагоприятен знак е възрастта над 50 години, както и развитието на аномалии във функционирането на човешкото тяло.
Сега знаете каква е същността на синдрома на ALS, както и на миелопатията. Защо възниква заболяването, какви форми и симптоми съществуват, както и какви методи на лечение се прилагат. Тъй като синдромът е смъртоносен, важно е да се покаже пациентът на специалист при първите признаци, за да се извърши необходимата терапия, за да се удължи и подобри живота на болния.

Смисълът на живота е свързан с въпроса „За какво да живеем“, а не с въпроса как да поддържаме живота. Отношението на човек е...

Гъбата е жив организъм, който образува отделно царство със същото име. Дълго време те са били класифицирани като част от растителното царство. Но в...

За любителите на „тихия“ лов сезонът на гъбите започва в началото на лятото и продължава до късна есен. И рядко го правят...

Алешникова, В.И. Използване на професионални консултанти. - М.: Инфра-М, 1999. - 240 с. 2. Бийч, Е....

Портокалов сок. Символичното значение на портокаловия сок в книгите за сънища е удоволствие и изкушение. Доста често ние...

Най-често преодоляваме всякакви трудности, срещани по пътя на живота. Разбира се, за това ние правим...

Тази година вашият покровител Нептун ще бъде във вашето съзвездие и това е добър знак, защото ще...

1993 кой? 1993 е годината на кое животно? — Според китайския хороскоп 1873, 1933, 1993 г. принадлежат към годините на Черния...

Двойствеността вълна-частица на светлината означава, че светлината едновременно има свойствата на непрекъснато...

Ролята на биологията е огромна в нашия свят. Въпреки че не е сред приоритетните предмети, повечето ученици и...
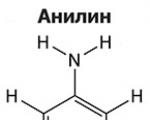
Амините са органични производни на амоняка, съдържащи NH2 аминогрупа и органичен радикал. Общо взето...
Как да отговаряте на въпроси в част B. Втората част от работата по социални науки се състои от 7 задачи с кратък отговор....

Формуляр TORG-15 се съставя в случай, когато по време на транспортиране, движение между и в склада, когато...

Диетолозите казват, че за добро здраве и стройна фигура трябва да включите леки закуски в...

Гъбата е жив организъм, който образува отделно царство със същото име. Дълго време те бяха класифицирани като кралство...

За любителите на „тихия“ лов сезонът на гъбите започва в началото на лятото и продължава до късна есен. И рядко го правят...