Visão filosófica do problema
O sentido da vida está relacionado com a questão “Para que viver”, e não com a questão de como manter a vida. A atitude de uma pessoa é...
Uma das doenças raras e extremamente perigosas para a saúde e a vida humana é a esclerose lateral amiotrófica. Esta é uma patologia do sistema nervoso em que os neurônios motores medula espinhal, assim como o córtex e o tronco cerebral, sofrem alterações irreversíveis. A doença é crônica e caracterizada por progressão constante. O processo patológico não é muito comum: são registrados aproximadamente 4 a 6 casos da doença por 100 mil pessoas.
A esclerose lateral amiotrófica (ELA) é um distúrbio no funcionamento do sistema nervoso que provoca fraqueza muscular, que ocorre sob condições de dano seletivo aos neurônios motores. A patologia tem vários outros nomes, por exemplo, doença de Lou Gehrig, em homenagem ao jogador campeão mundial de beisebol que sofria desse distúrbio. Outro nome para lado esclerose amiotrófica- Doença de Charcot. Está associado ao nome do psiquiatra francês Jean-Martin Charcot, que descreveu fraqueza muscular patológica na segunda metade do século XIX.
Finalmente, a ELA é conhecida como doença do neurônio motor de acordo com a CID-10.

Às vezes, ao descrever isso condição patológica, a palavra “lateral” é substituída pelo epíteto “lateral”. A inclusão desse adjetivo no nome da doença se deve ao fato de os neurônios localizados nas projeções laterais da medula espinhal serem os mais suscetíveis a alterações.
As alterações patológicas associadas ao enfraquecimento e atrofia muscular são causadas pela destruição de neurônios responsáveis pela transmissão de sinais do cérebro para os músculos. Com esta doença, os neurônios motores centrais e periféricos podem ser afetados. O primeiro deles está localizado no córtex hemisférios cerebrais cérebro Quando está danificado, desenvolve-se fraqueza muscular, enquanto o tônus aumenta e os reflexos ficam mais fortes. O neurônio motor periférico está localizado no tronco cerebral e em vários níveis medula espinhal. Se sofrer alterações patológicas, observa-se o desenvolvimento de fraqueza muscular, mas ao mesmo tempo os reflexos diminuem, assim como o tônus muscular, bem como o desenvolvimento de atrofia muscular.
Em condições de lesão de um dos neurônios motores (ou de ambos ao mesmo tempo), a transmissão de impulsos dele para o músculo é bloqueada.
A esclerose lateral amiotrófica manifesta-se pelo aumento da fraqueza muscular e da perda muscular, o que, como resultado, curso crônico, leva à imobilidade completa do paciente e ao comprometimento das funções respiratórias.

Atualmente, existem cerca de 70.000 pessoas no mundo diagnosticadas com ELA. Normalmente esta patologia se manifesta após os 40-50 anos de idade. Segundo dados clínicos, geralmente leva à morte dentro de 5 anos após o diagnóstico. No entanto, o famoso físico Stephen Hawking convive com esta doença há 50 anos.
Apenas 7% de todos os pacientes vivem mais de 60 meses.
Foi estabelecido que os homens sofrem da doença de Charcot com mais frequência do que as mulheres. Recentemente, os cientistas sugeriram que maior número casos de patologia são registrados em pessoas com alta inteligência e atletas que não tiveram problemas graves de saúde ao longo da vida.
A esclerose lateral amiotrófica tornou-se conhecida pela medicina há pouco tempo e a principal causa desta doença ainda não está clara. No entanto, os pesquisadores concordam que se baseia no acúmulo de proteínas patológicas insolúveis nas células motoras do sistema nervoso. Isto é o que leva à sua morte.
Existe uma teoria segundo a qual o papel de liderança do desenvolvimento mudanças irreversíveis refere-se a uma alteração nas propriedades de uma enzima especial, cuja função é proteger as células do corpo da destruição pelos radicais de oxigênio. Esta enzima é chamada superóxido dismutase-1. Tais alterações estão associadas a mutações genéticas, que em 25% dos casos são hereditárias. É esse fato que motiva a hipótese sobre natureza genética BAIXO.
É possível que a destruição dos neurônios motores esteja associada a mutações em outras estruturas, por exemplo, formações que dão forma à célula nervosa e fornecem sua estrutura.
Ao descrever a natureza da esclerose lateral amiotrófica, eles mencionam seguintes razões seu desenvolvimento:
Apresentadores do programa “Viver Saudável!” vou falar sobre doença incurável mais detalhes:
Os médicos incluem os seguintes fatores de risco:
A esclerose lateral amiotrófica não é doença infecciosa, eles não podem ser infectados por outra pessoa.
A doença do neurônio motor é classificada em tipos com base na gravidade dos danos às células nervosas. As manifestações de cada um deles são basicamente semelhantes, mas à medida que se desenvolvem processo patológico, a diferença se torna mais óbvia.
Na neurologia existem seguintes formulários doenças:

A foto mostra paralisia bulbar progressiva
A progressão da esclerose lateral amiotrófica causa complicações que levam à morte do paciente. Estes devem incluir:
A doença começa com danos nos membros, que depois se espalham para o resto do corpo.
Os primeiros sintomas da esclerose lateral amiotrófica são:
Caros leitores, sobre as principais causas e sintomas da doença, assista ao vídeo abaixo:
À medida que a esclerose lateral amiotrófica progride, o paciente percebe rigidez muscular, que está associada ao aumento do tônus e à dificuldade de tentar relaxá-los. Existem também tais características características, como desequilíbrio, crises espontâneas de riso ou choro, movimentos limitados da língua, alteração na voz.
Nos estágios posteriores da doença, ocorrem interrupções na respiração, são observadas depressão e incapacidade de se mover de forma independente.
O diagnóstico da esclerose lateral amiotrófica requer um grande número de medidas específicas. Em primeiro lugar, o paciente é examinado e entrevistado por um neurologista interessado nas seguintes informações:
Outros métodos que permitem fazer um diagnóstico correto incluem:

O procedimento para realizar uma punção lombar envolve perfurar a membrana aracnóide da medula espinhal entre a 3ª e a 4ª, ou 2ª e 3ª vértebras lombares com uma agulha de Beer para coletar o líquido cefalorraquidiano.
Além dos métodos listados, deve ser feito diagnóstico diferencial. A esclerose lateral amiotrófica deve ser diferenciada de patologias como tumor da medula espinhal, mielopatia cervical, síndrome de má absorção e amiotrofia diabética.
O tratamento da esclerose lateral amiotrófica visa retardar a progressão do processo patológico, uma vez que as alterações que ocorrem são irreversíveis. A doença não pode ser curada.
O principal e único medicamento atualmente recomendado para retardar a progressão da doença é o Riluzol, ou Rilutek. Seu principal princípio ativo evita maiores danos aos neurônios, retardando assim a progressão da doença.
Outros medicamentos são utilizados apenas para aliviar os sintomas predominantes que prejudicam a qualidade de vida do paciente. Estes devem incluir:

Diferentes tipos de ação dos relaxantes musculares
Um paciente com diagnóstico de ELA necessita de dispositivos que lhe permitam se movimentar (cadeira de rodas, andador, cama com várias funções, em particular, equipado com elevador). Para que o paciente possa respirar, pode ser realizada uma operação especial - traqueostomia. Durante a manipulação na traquéia cirurgicamente crie um buraco.
O paciente deve ser fornecido cuidado completo. Deve ser dada especial atenção às medidas para prevenir a formação de escaras. A cama deve estar sempre seca e limpa, assim como o corpo do paciente.
Outro componente integrante da terapia de manutenção para a esclerose lateral amiotrófica é trabalhar com um psicólogo. A ajuda de um especialista será necessária não só para o paciente, mas também para seus familiares.

Devido à incapacidade devido à ELA, o paciente é obrigado a usar uma cadeira de rodas
O prognóstico para esta doença específica é ruim. A doença progride constantemente, piorando cada vez mais a qualidade de vida do paciente e, em última análise, levando à insuficiência respiratória aguda e à morte do paciente. Dependendo da forma como ocorre o processo patológico, o paciente pode viver de 2 a 12 anos. Com a forma bulbar, assim como se o paciente for idoso, a expectativa de vida é reduzida para 1 a 3 anos.
A esclerose lateral amiotrófica é uma doença rara que progride constantemente e leva inevitavelmente à morte do paciente. As razões para este fenômeno não foram totalmente estudadas, existem apenas suposições sobre prováveis fatores de risco. O tratamento da ELA se resume a aliviar a condição do paciente e proporcionar-lhe as condições de vida mais confortáveis.
Esclerose lateral amiotrófica (ELA), também chamada de doença do neurônio motor ou Charcot-Kozhevnikov, doença do neurônio motor e, em alguns lugares do mundo, doença de Lou Gehrig, que afeta principalmente regiões de língua falada. língua Inglesa. Caros pacientes, a esse respeito, não devem se surpreender ou duvidar se no texto do nosso artigo se depararem com vários nomes para esse péssimo processo patológico, levando primeiro à incapacidade completa e depois à morte.
A base desta terrível doença são as lesões do tronco cerebral, que não param nesta área, mas se espalham para os cornos anteriores da medula espinhal (ao nível do espessamento cervical) e para os tratos piramidais, levando à degeneração dos músculos esqueléticos. Nas preparações histológicas, são encontradas inclusões citoplasmáticas chamadas corpos de Bunin e, no contexto de infiltrados vasculares, são observadas alterações degenerativas, enrugadas e mortas células nervosas, no lugar do qual crescem os elementos gliais. É óbvio que o processo, além de todas as partes do cérebro e da medula espinhal (cerebelo, tronco cerebral, córtex, subcórtex, etc.), núcleos motores dos nervos cranianos (nervos cranianos), afeta meninges, vasos cerebrais e leito vascular espinhal. Durante a autópsia, o patologista observa que o espessamento cervical e lombar dos pacientes está visivelmente reduzido em volume e o tronco está completamente atrofiado.

Se há 20 anos os pacientes mal conseguiam viver 4 anos, no nosso tempo tem havido uma tendência crescente duração média vida, que já chega aos 5-7 anos. A forma cerebral ainda não tem longevidade (3-4 anos), e a forma bulbar não oferece muitas chances (5-6 anos). É verdade que alguns vivem 12 anos, mas principalmente são pacientes com a forma cervicotorácica. Porém, o que significa esse período se a doença de Charcot (formas esporádicas) não poupa crianças (ensino médio) e adolescência, enquanto o sexo masculino tem maior “chance” de adquirir doença do neurônio motor. Os casos familiares aparecem com mais frequência na idade adulta. O perigo real de adoecer permanece entre os 40 e os 60 anos, mas depois dos 55 os homens já não lideram e adoecem tal como as mulheres.
Distúrbios bulbares na atividade dos centros responsáveis pela função e trabalho respiratório geralmente levam à morte. do sistema cardiovascular.
Na literatura você pode encontrar uma definição como “síndrome de ELA”. Esta síndrome não tem nada a ver com doença do neurônio motor, é causada por razões completamente diferentes e acompanha outras doenças (algumas proteinemia, etc.), embora os sintomas da síndrome de ELA lembrem muito o estágio inicial da doença de Lou Gehrig, quando a clínica ainda não se desenvolveu rapidamente. Pela mesma razão Estado inicial A esclerose lateral amiotrófica é diferenciada de () ou.
 A ELA não tem limites no corpo humano doente, ela avança e, assim, atinge todo o corpo do paciente, portanto as formas de esclerose lateral amiotrófica são diferenciadas de forma bastante condicional, com base no início do processo patológico e mais sinais claros derrotas. Exatamente predominante os sintomas da esclerose lateral amiotrófica, e não as áreas afetadas isoladas, permitem determinar suas formas, que podem se apresentar da seguinte forma:
A ELA não tem limites no corpo humano doente, ela avança e, assim, atinge todo o corpo do paciente, portanto as formas de esclerose lateral amiotrófica são diferenciadas de forma bastante condicional, com base no início do processo patológico e mais sinais claros derrotas. Exatamente predominante os sintomas da esclerose lateral amiotrófica, e não as áreas afetadas isoladas, permitem determinar suas formas, que podem se apresentar da seguinte forma:
Os fatores que podem desencadear esse grave processo patológico não são tão numerosos, mas uma pessoa pode enfrentar qualquer um deles todos os dias, independentemente da idade, sexo e localização geográfica, exceto, é claro, a predisposição hereditária, típica apenas de uma determinada parte da população (5-10%).
Então, as causas da doença do neurônio motor:
Os sintomas da esclerose lateral amiotrófica são caracterizados principalmente pelo aparecimento de lesões periféricas e paresia central mãos, conforme indicado pelos seguintes sinais:

É óbvio que envolvendo todo o corpo no processo, a doença de Charcot apresenta sintomas ricos e diversos, que, no entanto, podem ser brevemente representados por síndromes:
Quanto ao diagnóstico, depende principalmente do estado neurológico, e o principal método instrumental A ENMG (eletroneuromiografia) é reconhecida para pesquisar ELA; outros exames são realizados para excluir doenças com sintomas semelhantes ou para estudar o corpo do paciente, em particular, o estado do sistema respiratório e do sistema músculo-esquelético. Assim, a lista pesquisa necessária inclui:
A terapia para doenças do neurônio motor é principalmente destinado a fortalecimento geral, mantendo o corpo e aliviando os sintomas.À medida que o processo patológico se desenvolve, a insuficiência respiratória aumenta, portanto, para melhorar a atividade respiratória, o paciente primeiro (ainda na cadeira de rodas) muda para um dispositivo de VNI (para ventilação não invasiva dos pulmões) e depois, quando puder não conseguem mais lidar com equipamentos de ventilação estacionários.

Sério remédio eficaz ainda não foi inventado para o tratamento da esclerose lateral amiotrófica, entretanto, o tratamento ainda é necessário e o paciente recebe terapia medicamentosa:
Dificilmente se pode contestar a afirmação de que um paciente com doença de Charcot necessita de cuidados especiais. É especial, porque só a alimentação já vale a pena. E a luta contra as escaras? E a depressão? O paciente critica seu estado, fica muito preocupado porque a cada dia seu quadro piora e, no final das contas, ele para (não por vontade própria) de cuidar de si mesmo, não consegue se comunicar com os outros e saborear um delicioso jantar.
Esse paciente precisa de:
A prevenção de escaras é muito importante. Eles estão dentro casos semelhantes não fique esperando muito, por isso a cama deve estar limpa e seca, assim como o corpo do paciente.
O paciente ingere principalmente alimentos líquidos e de fácil ingestão, rico em proteínas e vitaminas (desde que a função de deglutição seja preservada). Posteriormente, o paciente é alimentado por sonda, e então recorrem a uma medida forçada, mas última - a imposição de gastrostomias.

É óbvio que um paciente com esclerose lateral amiotrófica sofre muito: tanto moral quanto fisicamente. Ao mesmo tempo, as pessoas que cuidam dele, de quem ele é uma pessoa próxima, também sofrem. Concordo, é muito difícil olhar nos olhos desbotados, ver a dor e o desespero e não poder ajudar a derrotar a doença, curá-la, trazê-la de volta à vida. querida pessoa. Os parentes que cuidam de uma pessoa tão doente perdem as forças e muitas vezes ficam desanimados e estado depressivo, e por isso também necessitam do auxílio de um psicoterapeuta com prescrição de sedativos e antidepressivos.
Normalmente, na descrição do tratamento de qualquer doença, o leitor procura medidas preventivas e formas de se livrar de uma determinada enfermidade. remédios populares. Na verdade recomendado Medicina alternativa, contendo grandes quantidades de vitaminas B, gérmen de trigo e grãos de aveia, nozes e a própolis pode não fazer mal ao paciente, mas também não vai curá-lo. Além disso, não devemos esquecer que essas pessoas têm frequentemente problemas de deglutição, por isso no caso da doença de Charcot, não se deve confiar na medicina tradicional.
Isto é o que é - esclerose lateral amiotrófica (e muitos outros nomes para ela). A doença é terrivelmente insidiosa, incompreensível e incurável. Talvez um dia uma pessoa consiga domar esta doença, pelo menos, esperemos o melhor, porque cientistas de todo o mundo estão trabalhando neste problema.
A esclerose lateral amiotrófica (ELA) é uma doença incurável e progressiva do sistema nervoso central, na qual o paciente sofre danos nos neurônios motores superiores e inferiores, o que causa atrofia muscular e paralisia. A frequência desta patologia é de cerca de 2 a 7 casos por 100 mil pessoas. Na maioria das vezes, a doença é diagnosticada em pacientes com mais de 50 anos de idade.
Os cientistas ainda não criaram uma classificação unificada e abrangente de ELA. Existem várias abordagens para classificar a doença. Por exemplo, a abordagem norte-americana envolve a identificação dos seguintes tipos de ELA: esporádica, familiar, endêmica esporádica. A classificação da esclerose lateral amiotrófica prevê as seguintes formas da doença: bulbar, lombossacra, cervicotorácica e primária generalizada. Existem também diversas variantes da doença: mista, piramidal e segmentar-nuclear.
Para o mais comum sintomas iniciais As doenças incluem cãibras (espasmos musculares dolorosos), letargia e fraqueza nos braços distais, distúrbios bulbares, atrofia muscular das pernas, fraqueza nos cintura escapular. Além disso, para opções diferentes A doença é caracterizada por diversas manifestações clínicas.
As causas exatas da esclerose lateral amiotrófica ainda estão sendo investigadas pelos cientistas. Porém, podem ser citados vários fatores que provocam a doença. Por exemplo, cerca de 5% das doenças têm etiologia hereditária. Pelo menos 20% dos casos estão associados a mutações no gene da superóxido dismutase-1. Os cientistas provaram que papel importante A alta atividade do sistema glutamatérgico desempenha um papel no aparecimento da doença. O fato é que o excesso de ácido glutâmico provoca superexcitação e morte súbita dos neurônios. O mecanismo genético molecular da patologia também foi comprovado. É causada por um aumento no nível de DNA e RNA nas células, o que acaba levando à interrupção da síntese protéica.
Os cientistas também identificam vários fatores predisponentes que desempenham um papel importante na ocorrência de ELA. Em primeiro lugar, esses fatores incluem a idade. O fato é que a doença geralmente se desenvolve em pacientes com idade entre 30 e 50 anos. Vale lembrar que apenas cerca de 5% dos pacientes apresentam predisposição hereditária para ELA. Na grande maioria dos casos de ELA, a causa da patologia não pode ser determinada.
O curso inicial da doença é caracterizado por sintomas como convulsões, espasmos, dormência muscular, dificuldade para falar e fraqueza nos membros. Como esses sintomas são característicos de muitas doenças neurológicas, é difícil diagnosticar ELA em um estágio inicial. Na maioria dos casos, a doença pode ser diagnosticada na fase de atrofia muscular.
Dependendo da doença afetada partes diferentes corpo, distinguir ELA de membros e ELA bulbar. No primeiro caso, os pacientes apresentam deterioração da flexibilidade do tornozelo, falta de jeito ao caminhar e começam a tropeçar. A ELA bulbar se manifesta por dificuldade para falar (som nasal, dificuldade para engolir). Logo o paciente sente dificuldade para se movimentar ou não consegue mais se movimentar de forma independente. Geralmente a doença não tem um efeito prejudicial sobre capacidade mental paciente, mas leva à depressão grave. Na maioria dos casos, decorrem cerca de três a cinco anos desde o aparecimento dos primeiros sintomas até à morte.
Como a ELA é uma doença incurável que encurta rapidamente a vida de uma pessoa, o exame do paciente deve ser abrangente e preciso. É extremamente importante colocar diagnóstico correto o paciente para iniciar o alívio oportuno de seus principais sintomas, pois isso pode prolongar a vida do paciente. O plano de exame geralmente inclui história de vida e doença, exame neurológico e físico, ressonância magnética da medula espinhal e do cérebro, EMG e exames laboratoriais.
O diagnóstico da doença começa com uma entrevista detalhada com o paciente. Ou seja, o médico precisa esclarecer se o paciente se queixa de espasmos e espasmos musculares, fraqueza e rigidez, dificuldade de movimento das mãos, fala, caminhada, deglutição, salivação, falta de ar frequente, perda de peso, fadiga, falta de ar durante o exercício. Além disso, o médico deve perguntar se o paciente notou visão dupla, perda de memória, sensação de formigamento pelo corpo ou problemas urinários. É imprescindível perguntar ao paciente sobre sua história familiar - se ele tem parentes com distúrbios crônicos do movimento.
O principal objetivo do exame físico é avaliar a constituição do paciente, pesá-lo, medir sua altura e calcular seu índice de massa corporal. O exame neurológico geralmente inclui testes neuropsicológicos. Ao avaliar as funções bulbares, o médico atenta para o timbre da voz, velocidade da fala, reflexo faríngeo, presença de atrofias da língua e paresia do palato mole. Além disso, durante o exame, é verificada a força dos músculos trapézios.
O principal método instrumental para diagnosticar a doença é a EMG de agulha. Essa técnica permite identificar sinais da doença, como desnervação aguda ou crônica. Nos estágios iniciais da doença, a estimulação EMG é ineficaz porque não detecta sinais perceptíveis de ELA.
No processo de diagnóstico da doença, os médicos também utilizam métodos de neuroimagem. A ressonância magnética da medula espinhal e do cérebro desempenha um grande papel no diagnóstico diferencial da ELA. Durante a ressonância magnética, em 17-67% dos pacientes é possível identificar sintomas de degeneração dos tratos piramidais e atrofia do córtex motor do cérebro. Porém, vale ressaltar que esta técnica é ineficaz no diagnóstico da doença em pacientes com síndrome bulbar.
Muitos exames laboratoriais são realizados durante o diagnóstico de ELA. Em particular, os médicos podem prescrever medicamentos clínicos e testes bioquímicos sangue, exame do líquido cefalorraquidiano, estudos sorológicos. No entanto, o único método de análise eficaz e confiável ainda é considerado a análise genética molecular. A presença de mutações no gene da superóxido dismutase-1 é considerada suspeita de ELA.
Como os sintomas da esclerose lateral amiotrófica são em muitos aspectos semelhantes às manifestações de outras patologias neurológicas, os médicos devem realizar diagnóstico diferencial. O diagnóstico mais preciso pode ser feito por meio de ressonância magnética do cérebro e da coluna. Em primeiro lugar, a ELA deve ser diferenciada das doenças musculares, que incluem a miotonia distrófica de Rossolimo-Steinert-Kurshman, a miosite com anomalias celulares e a miodistrofia oculofaríngea.
Também é necessário distinguir a ELA das patologias da medula espinhal:
O diagnóstico diferencial também é necessário para distinguir a doença de patologias sistêmicas, lesões da sinapse neuromuscular e patologias cerebrais, como atrofia de múltiplos sistemas, encefalopatia discirculatória e siringobulbia.
Considera-se que os principais objetivos do tratamento da esclerose lateral amiotrófica são retardar a progressão da doença, bem como eliminar seus sintomas, que pioram significativamente a qualidade de vida do paciente. Deve-se lembrar que a ELA é uma doença grave e incurável que reduz a expectativa de vida de uma pessoa. É por isso que o médico tem o direito de informar o paciente sobre o diagnóstico somente após um exame completo e minucioso.
O tratamento da doença inclui terapia medicamentosa e não medicamentosa. Este último implica medidas de segurança. O paciente deve limitar a atividade física, o que pode acelerar a progressão da ELA. Além disso, é muito importante comer de forma adequada e nutritiva. A terapia medicamentosa é dividida em dois tipos: patogenética e paliativa.
Até o momento, o único medicamento que pode retardar a progressão da ELA é o riluzol. Está comprovado que tomá-lo pode prolongar a vida do paciente em média três meses. Este medicamento é indicado para pacientes com duração da doença inferior a 5 anos. O paciente deve receber 100 mg do medicamento diariamente. Para evitar o risco de hepatite induzida por drogas, a cada três meses é necessário verificar os níveis de AST, ALT e LDH. Como os homens e os fumadores têm concentrações mais baixas de riluzol no sangue, devem limitar o consumo de tabaco ou deixar de fumar completamente. mau hábito. Você precisará tomar o medicamento pelo resto da vida.
Os cientistas tentaram repetidamente usar outros medicamentos para terapia patogenética. No entanto, tais experimentos não se mostraram eficazes. Entre eles estavam:
A eficácia de tomá-lo também não foi comprovada altas doses Cerebrolisina, apesar de este medicamento ser capaz de melhorar ligeiramente o estado dos pacientes.
A terapia paliativa visa eliminar um complexo de sintomas da doença e, assim, melhorar a qualidade de vida do paciente. Para eliminar certos sintomas da ELA, são utilizadas as seguintes técnicas:
Para melhorar o metabolismo muscular, um paciente com ELA pode receber os seguintes medicamentos: creatina, carnitina, solução de levocarnitina, propionato de trimetilhidrazínio. A terapia multivitamínica também é indicada para pacientes, o que envolve o uso de multivitaminas (neuromultivit, milgamma) e ácido tióctico.
Na maioria dos pacientes com ELA, a doença é acompanhada por graves deficiências motoras, incluindo mobilidade limitada. É claro que isso causa grande desconforto ao paciente, que precisa constantemente da ajuda de outras pessoas. Elimine alguns distúrbios do movimento As técnicas de correção ortopédica ajudam. O médico deve explicar ao paciente que o uso de dispositivos assistivos não indica sua deficiência, mas apenas reduz as dificuldades causadas pela doença.
O sintoma mais fatal da doença é considerado insuficiência respiratória. Seus primeiros sintomas serão fadiga matinal, sonhos vívidos, sonolência diurna e insatisfação com o sono. Para detectar insuficiência respiratória precocemente, são realizadas polissonografia e espirografia. Para eliminar a apneia, são indicados medicamentos e ventilação não invasiva. Está comprovado que essas técnicas podem prolongar a vida de um paciente em um ano. Se o paciente necessitar de respiração assistida por mais de 20 horas, o médico levanta a questão de uma transição completa para ventilação invasiva.
Pacientes que tenham sido submetidos a um exame inicial ou a um diagnóstico repetido da doença devem permanecer sob observação ambulatorial. À medida que surgem novos sintomas, eles também devem receber aconselhamento qualificado. Os pacientes devem tomar a maioria dos medicamentos regularmente. Apenas vitaminas e medicamentos miotrópicos são administrados em etapas.
A cada três meses o paciente deve realizar espirografia. Se ele toma riluzol regularmente, ele precisa verificar LDH, AST e ALT a cada seis meses. Se o paciente apresentar disfagia, os níveis de glicose no sangue e o estado trófico devem ser medidos periodicamente. Os pacientes podem escolher o regime de tratamento: podem ficar em casa ou em um hospício.
O prognóstico para pacientes com ELA depende em grande parte do curso da doença. Está comprovado que cerca de 80-90% dos pacientes que apresentam complicações respiratórias graves morrem dentro de 3-5 anos após o aparecimento dos primeiros sinais da doença. Os 10% restantes dos pacientes apresentam um curso benigno da doença. A duração da doença é significativamente reduzida na presença dos seguintes fatores: idade do paciente inferior a 45 anos, início bulbar da ELA, rápida progressão da doença.
A esclerose lateral amiotrófica é uma doença neurológica grave que causa fraqueza muscular, incapacidade e, por fim, morte. A ELA é frequentemente chamada de doença de Lou Gehrig, em homenagem ao famoso jogador de beisebol que foi diagnosticado em 1939. Em alguns países, a ELA e a doença do neurónio motor são por vezes utilizadas de forma intercambiável.
Em todo o mundo, a ELA afeta 1-3 pessoas em cada 100.000. Na grande maioria dos casos - de 90 a 95 por cento dos casos desta doença, os médicos não conseguem explicar a causa da doença. Apenas em 5 a 10 por cento dos casos a determinação genética é rastreada. A ELA geralmente começa com cãibras musculares em um braço ou perna e dificuldade para falar. Em última análise, a ELA perturba o controle dos músculos responsáveis pelos movimentos respiratórios da deglutição.
Os primeiros sintomas da ELA incluem:
A doença geralmente começa nos braços, pernas ou membros e depois se espalha para outras partes do corpo. Com o início da doença, os sintomas começam a progredir, os músculos ficam mais fracos e ocorre a paralisia. Em última análise, há uma violação dos atos de mastigar, engolir e respirar.
Na esclerose lateral amiotrófica, as células nervosas que controlam o movimento (neurônios motores) começam a morrer gradualmente, o que leva a um enfraquecimento gradual dos músculos e à sua atrofia. ALS é herdada em 5 a 10 por cento dos casos. Em outros casos, a ELA parece ocorrer espontaneamente.
Várias possíveis causas de ELA estão sendo estudadas atualmente, incluindo:
Os principais fatores de risco para ELA incluem:
Os fatores ambientais que aumentam o risco desta doença incluem:
 À medida que a doença progride, os pacientes com esclerose lateral amiotrófica apresentam as seguintes complicações.
À medida que a doença progride, os pacientes com esclerose lateral amiotrófica apresentam as seguintes complicações.
Caso apareçam alguns dos primeiros sintomas de doenças neuromusculares, deve contactar o seu médico que, se necessário, o encaminhará para uma consulta com um neurologista. Mas mesmo uma visita oportuna a um neurologista não garante que o diagnóstico seja feito imediatamente, pois leva algum tempo para verificar o diagnóstico. O neurologista estará interessado no histórico médico e no estado neurológico.
A esclerose lateral amiotrófica é bastante difícil de diagnosticar nas fases iniciais, uma vez que os sintomas são semelhantes a outros doenças neurológicas. Os seguintes métodos de diagnóstico são usados:
Devido ao fato de os processos da esclerose lateral amiotrófica não poderem ser revertidos, o tratamento visa retardar a progressão dos sintomas.
Tratamento medicamentoso. O medicamento riluzol (RILUTEK) é o primeiro e único medicamento aprovado para retardar a ELA. O medicamento tem efeito inibitório na progressão da doença em alguns pacientes, possivelmente por redução do nível de glutamato, substância que é mediadora do sistema nervoso e cujo nível costuma estar elevado em pacientes com ELA. Além disso, outros medicamentos podem ser prescritos para aliviar sintomas como prisão de ventre. cãibras musculares fadiga hipersalivação dor depressão.
Terapia por exercício. Os exercícios físicos sob a supervisão de um fisioterapeuta podem ajudar a manter a força muscular e a amplitude de movimento por mais tempo. um longo período atividade do sistema cardiovascular e melhorar o bem-estar geral.
Usando um andador ou cadeira de rodas também permite que você mantenha uma certa amplitude de movimentos.
Ajuda psicológica. A ajuda de um psicólogo muitas vezes é necessária devido à consciência do paciente sobre a incurabilidade da doença. Embora em alguns casos a expectativa de vida possa ultrapassar 3-5 anos e chegar a 10 anos.
Síndrome de ELA significa. A doença afeta o sistema nervoso central, desenvolve-se lentamente, mas ataca a medula espinhal e o cérebro, bem como os núcleos dos nervos cranianos. Se não for tratada, a doença pode danificar os neurônios motores, causando paralisia e atrofia muscular. Vejamos o que é mielopatia e quais são seus sintomas, causas e como o tratamento é realizado.
A mielopatia é uma patologia da medula espinhal que pode se desenvolver na síndrome de ELA. Se o funcionamento da medula espinhal for perturbado, as funções e reações forem insuficientes, a pessoa enfrentará consequências terríveis.
Razões que provocam o aparecimento de mielopatia:
A doença pode se desenvolver devido a um desses fatores ou a vários.
A mielopatia apresenta os seguintes sintomas:

Os sintomas listados requerem terapia. Mas antes de iniciar o tratamento, é importante diagnosticar a mielopatia. Equipamentos modernos e testes diferenciais são utilizados para fazer o diagnóstico.
A mielopatia requer tratamento. Doença em período agudo tratado eliminando a dor na área afetada. O tratamento é realizado por meio de bloqueio. A pele ao redor da lesão é injetada com analgésicos. Graças ao bloqueio, o cérebro não recebe sinal sobre a presença de inflamação nas articulações ou músculos. Como resultado, a dor fica bloqueada por muito tempo.
Depois disso, o tratamento é realizado com:
Graças a essas medidas, o bem-estar do paciente melhora.
Muitas vezes a mielopatia evolui para forma crônica. Nesse caso, para evitar que a doença progrida e se agrave, é importante mantê-la em estado de remissão.
Para que a doença incomode o mínimo possível, é importante fazer a sua prevenção.
As medidas preventivas visam prevenir as causas que provocam a diminuição das funções da medula espinhal. 
Você precisa comer direito, endurecer, tentar se livrar excesso de peso. Para prevenir qualquer doença, fortaleça seu sistema imunológico em todos os sentidos.
Para as crianças, para prevenir doenças, crie um horário de trabalho e descanso e distribua a carga.
Devido à mutação de proteínas com o desenvolvimento de agregados intracelulares, a doença ELA começa a se formar. A doença geralmente afeta pessoas entre 40 e 60 anos de idade.
As causas exatas do desenvolvimento da doença não são totalmente compreendidas. Mas os cientistas descobriram que a doença se desenvolve devido ao aparecimento de DNA de quatro cadeias nas células humanas. Como resultado, a síntese protéica é interrompida e os sintomas da síndrome começam a aparecer.
Para fazer um diagnóstico preciso, é importante consultar vários especialistas.
5% das pessoas adquirem a doença de parentes devido a um fator hereditário.
As causas da síndrome podem ser infecções, lesões ou doenças infecciosas.
A maioria das pessoas hoje apresenta sintomas da síndrome de ELA. Em cada caso, a doença tem suas próprias causas e sintomas de desenvolvimento. É importante monitorar sua saúde para identificar a patologia nos estágios iniciais de manifestação e iniciar seu tratamento. 
Sintomas da síndrome:
A doença de ELA é muito difícil de diagnosticar nos estágios iniciais. Mas especialistas experientes estudam os sintomas, consideram versões diferentes doenças, e só depois é feito o diagnóstico e prescrito o tratamento.
Mesmo assim, se o paciente for diagnosticado com ELA, seus familiares precisam se preparar para as dificuldades.
O paciente perde completamente a capacidade de se mover de forma independente. Então ele não pode comer normalmente. Também às vezes há salivação muito forte. Alguns pacientes necessitam de nutrição enteral especial.
Depois de algum tempo, começam os problemas do sistema respiratório e ocorre insuficiência respiratória.
Os pacientes costumam ser incomodados por dores de cabeça, sufocamento e pesadelos. 
Levando em consideração a localização dos músculos, os especialistas identificam as seguintes formas de síndrome de ELA:
A forma bulbar da doença é caracterizada pelos seguintes distúrbios: dormência da língua, paralisia do palato, fraqueza dos músculos mastigatórios. A fala e a capacidade de engolir normalmente também ficam prejudicadas. EM Casos severos o paciente pode começar a rir ou chorar sem motivo, mover-se animadamente maxilar inferior. Depois de algum tempo, os braços e as pernas são afetados. Na maioria das vezes, mesmo após tratamento adequado, os pacientes morrem.
Na forma cervicotorácica da doença, os músculos dos braços e pernas atrofiam.
Na forma lombossacral, ocorre paresia atrófica das extremidades inferiores. Quando a forma avança, os braços e os músculos cranianos também ficam paralisados.
Na forma cerebral, os membros ficam paralisados e os neurônios motores periféricos são afetados. O paciente ri e chora sem motivo. Às vezes, ele começa a mover ativamente o maxilar inferior.
Para fazer um diagnóstico, é realizado um eletromonograma. O estudo mostra que existe um ritmo de “cerca de estacas” nos potenciais de fasticulação; a velocidade de condução não muda. Para fazer um diagnóstico correto é importante acompanhar todas as áreas da coluna:

É importante não confundir a síndrome com outros tipos de doenças que podem apresentar sintomas semelhantes. Portanto, um diagnóstico preciso deve ser feito por especialistas experientes.
O tratamento para ELA visa aliviar os sintomas da doença. A terapia é realizada com Riluzona. Porém, este medicamento está disponível apenas na Europa e nos EUA. A medicina não cura a doença. Mas ajuda a prolongar e melhorar a qualidade de vida de uma pessoa doente.
O tratamento da síndrome de ELA é realizado com os seguintes medicamentos:
O trabalho de Riluzon. Quando é transmitido? impulso nervoso, o glutamato, um mediador químico do sistema nervoso central, é liberado. Rizulon reduz a quantidade de excreção dessa substância.
Após pesquisas, os cientistas descobriram que o excesso de glutamato danifica os neurônios da medula espinhal e do cérebro. De acordo com testes, as pessoas que usam Rizulon vivem mais do que outros pacientes - três meses.
Os cientistas também descobriram que os antioxidantes suprimem os sinais da síndrome. Essas substâncias ajudam o corpo a prevenir os danos dos radicais livres. Os antioxidantes são selecionados pelo médico levando em consideração o estado de saúde do paciente. 
Para facilitar a vida das pessoas com a síndrome, tratamento concomitante. Como a doença demora muito para ser tratada, é importante tratar não só a doença principal, mas também outros sintomas. Segundo especialistas, o relaxamento alivia a ansiedade e o medo.
Reflexologia, aromaterapia e massagem são usadas para relaxar os músculos. Graças a esses procedimentos, a circulação linfática e sanguínea é normalizada e a dor é eliminada. Ao realizar todos os procedimentos, os especialistas estimulam analgésicos e endorfinas endógenas. Mas é importante realizar atividades com sistema nervoso individualmente. Portanto, antes de iniciar o tratamento, consulte um médico e faça todos os exames.
A síndrome progride constantemente e se não for tratada pode ser fatal. Uma vez detectados os sinais da doença, o paciente tem a oportunidade de viver mais cinco anos. Mas para que sua vida prossiga bem, forneça terapia de suporte.
Um sinal desfavorável é a idade superior a 50 anos, bem como o desenvolvimento de anomalias no funcionamento do corpo humano.
Agora você sabe qual é a essência da síndrome de ELA, assim como da mielopatia. Por que ocorre a doença, quais as formas e sintomas existentes e também quais métodos de terapia são realizados. Como a síndrome é mortal, é importante levar o paciente ao especialista aos primeiros sinais para realizar a terapia necessária para prolongar e melhorar a vida do doente.

O sentido da vida está relacionado com a questão “Para que viver”, e não com a questão de como manter a vida. A atitude de uma pessoa é...

Um cogumelo é um organismo vivo que forma um reino separado com o mesmo nome. Por muito tempo foram classificados como parte do reino vegetal. Mas em...

Para os amantes da caça “tranquila”, a temporada dos cogumelos começa no início do verão e vai até o final do outono. E raramente o fazem...

Aleshnikova, V.I. Uso de consultores profissionais. - M.: Infra-M, 1999. - 240 p. 2. Beich, E....

Suco de laranja. O significado simbólico do suco de laranja nos livros de sonhos é prazer e tentação. Muitas vezes nós...

Na maioria das vezes superamos todos os tipos de dificuldades encontradas ao longo do caminho da vida. Claro, para isso fazemos...

Este ano o seu patrono Netuno estará na sua constelação e isso é um bom sinal, pois você...

1993 quem? 1993 é o ano de qual animal? — De acordo com o horóscopo chinês, 1873, 1933, 1993 pertenciam aos anos do Negro...

A dualidade onda-partícula da luz significa que a luz tem simultaneamente as propriedades de continuidade...

O papel da biologia é enorme em nosso mundo. Embora não seja uma das disciplinas prioritárias, a maioria dos escolares e...
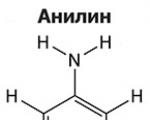
As aminas são derivados orgânicos da amônia contendo um grupo amino NH 2 e um radical orgânico. Em geral...
Como responder às perguntas da Parte BA segunda parte do trabalho de estudos sociais consiste em 7 tarefas com uma resposta curta....

O formulário TORG-15 é elaborado no caso de durante o transporte, movimentação entre e dentro do armazém, quando...

Os nutricionistas dizem que para ter uma boa saúde e um corpo esguio é preciso incluir lanches na sua...

Um cogumelo é um organismo vivo que forma um reino separado com o mesmo nome. Durante muito tempo foram classificados como um reino...

Para os amantes da caça “tranquila”, a temporada dos cogumelos começa no início do verão e vai até o final do outono. E raramente o fazem...