O que é possível e o que não é possível durante o Jejum da Natividade?
Em 2018, o Jejum da Natividade começará em 28 de novembro. Durante este período, os crentes ortodoxos se preparam para celebrar o Natal...
A policitemia verdadeira ou primária (policitemia vera) é uma doença tumoral crônica sistema circulatório pessoa. Expresso em salto brusco nível de glóbulos vermelhos no sangue humano sem danos órgãos internos. O sangue torna-se mais viscoso e espesso, o que leva à desaceleração do fluxo sanguíneo e, consequentemente, à formação de coágulos sanguíneos e hipóxia, que atinge em maior medida o germe eritroblástico. Esta doença foi descrita pela primeira vez por Váquez em 1892, por isso também é chamada de doença de Váquez.
Esta doença geralmente afeta pessoas cada vez mais idosas, e os homens são afetados com mais frequência. As crianças e os jovens adoecem com muito menos frequência, mas a sua doença é mais grave. As verdadeiras razões doenças não foram estabelecidas, mas foi comprovado predisposição genética para esta doença.
A doença se desenvolve lentamente; por algum tempo o paciente pode nem suspeitar que tem a doença, pois estágios iniciais não há sinais. Então, à medida que a doença progride, os pacientes começam a sentir tonturas, uma sensação incompreensível de crânio cheio e dores de cabeça constantes.
Na fase avançada da doença, são observados os seguintes sintomas clínicos:

Os pacientes também reclamam fadiga, deficiência visual, deficiência auditiva, instabilidade de humor e letargia geral. Além disso, a doença está associada a hipertensão, perda grave de peso, resfriados e bronquite.
O diagnóstico é feito após avaliação completa do quadro clínico, análise das alterações no sangue e exame medula óssea. Os seguintes indicadores são levados em consideração para confirmar a presença da doença de Vaquez.
O nível de glóbulos vermelhos e hemoglobina aumenta significativamente, pelo contrário, diminui e é inferior a um. Uma contagem elevada de glóbulos vermelhos leva a um aumento no volume total de sangue no corpo (até 2 vezes), e o hematócrito também muda (até 65%).
Os leucócitos aumentam 1,5–2 vezes, os neutrófilos crescem (até 80%). O número de plaquetas aumenta significativamente (mais de 400*109 por litro). A viscosidade do sangue aumenta significativamente, os glóbulos vermelhos não se depositam mais do que 2 milímetros por hora. A taxa também está aumentando rapidamente ácido úrico.
Usando trepanobiópsia, é feita uma punção da medula óssea. O exame histológico do puntiforme revela aumento de megacariócitos. Ao mesmo tempo, as células da medula óssea mantêm a capacidade de amadurecer no mesmo nível.
Há também a necessidade de produzir diagnóstico diferencial com eritrocitose secundária.
A doença de Vaquez tende a ocorrer na forma de um processo crônico prolongado. A vida do paciente está em risco risco aumentado sangramento e coágulos sanguíneos.
A principal direção do tratamento é combater a trombose e reduzir a viscosidade do sangue, que depende do nível de glóbulos vermelhos. Para tanto, são utilizadas sangrias e quimioterapia.
Normaliza o nível de hemoglobina e glóbulos vermelhos, reduzindo o volume sanguíneo do paciente. Volume – até 500 mililitros por procedimento. Tais procedimentos são realizados sob a supervisão de um hematologista uma vez a cada dois dias até que a hemoglobina (140–150) e o hematócrito (45%) diminuam dentro dos limites normais. Este método Estimula bem a medula óssea e alivia a coceira na pele. Na maioria dos casos, a sangria é usada em combinação com quimioterapia.
Usado para inibir a recuperação de glóbulos vermelhos e plaquetas. Para isso são usados preparações farmacológicas grupos diferentes: imifos, mielobromol, mielosan, clorambucil, e também usam hidroxiureia e pipobroman.
Policitemia vera(eritremia, doença de Vaquez, policitemia rubra) - IP é uma doença mieloproliferativa neoplásica crônica com lesão de células-tronco, proliferação de três linhagens hematopoiéticas, aumento da formação de hemácias e, em menor extensão, de leucócitos e plaquetas. Em determinado estágio da doença, ocorre metaplasia mieloide do baço.
A incidência de policitemia vera é de aproximadamente 1 aleatório por 100 mil habitantes por ano e em últimos anos tem uma tendência indubitável de aumentar. Os homens adoecem com um pouco mais de frequência do que as mulheres (1.2:1). Idade Média os doentes têm 60 anos, os pacientes com menos de 40 anos representam apenas 5%.
Etiopatogenia. A policitemia vera é uma doença neoplásica clonal, que se baseia na transformação de uma célula-tronco hematopoiética. Como a transformação maligna ocorre ao nível de uma célula-tronco pluripotente, todas as três linhagens de hematopoiese estão envolvidas no processo. Em pacientes que sofrem de PV, há aumento do conteúdo de UFC-GEMM (unidades formadoras de colônias - granulocíticas, eritróides, macrófagos e megacariócitos) - células progenitoras próximas a uma célula-tronco pluripotente. Na cultura celular, a proliferação activa destas células ocorre na ausência de eritropoietina. Níveis séricos baixos de eritropoetina são um sinal específico de PV. Na medula óssea, observa-se hiperplasia de células predominantemente eritróides, bem como de linhagens granulocíticas e megacariocíticas. Uma característica é a presença de aglomerados de megacariócitos polimórficos (de pequenos a gigantes). A mielofibrose raramente é observada no momento do diagnóstico, mas se manifesta claramente com um longo curso da doença. Gradualmente há um aumento no número de reticulina e fibras de colágeno, desenvolve-se mielofibrose e a mielopoiese é reduzida. A massa de eritrócitos circulantes (MCE) aumenta, o hematócrito aumenta, a viscosidade do sangue aumenta (há um aumento significativo no conteúdo de hemoglobina no sangue (de 180 g/l e acima), glóbulos vermelhos (de 6,6 x 10 12 /l) e o índice de hematócrito (de 55% e superior). Esses fatores, juntamente com a trombocitose, levam à microcirculação prejudicada e complicações tromboembólicas. Paralelamente, a metaplasia mieloide do baço está associada. Na IP não há marcador citogenético específico, entretanto, em um número significativo de pacientes com PI na fase de desenvolvimento de mielofibrose, anomalias cromossômicas.
Quadro clínico muda com o curso da doença e é determinado principalmente pelo estágio da doença. Na literatura nacional, costuma-se distinguir quatro estágios da IP, que refletem os processos patológicos que ocorrem na medula óssea e no baço dos pacientes
Estágios:
I - inicial, pouco sintomático (5 anos ou mais):
o baço não é palpável
eritrocitose moderada
abundância moderada
na panmielose da medula óssea
Complicações vasculares e trombóticas são possíveis, mas não são comuns
As manifestações externas da doença são pletora, acrocianose, eritromelalgia (dor em queimação, parestesia nas pontas dos dedos) e coceira na pele após a lavagem. Um aumento no MCE e, consequentemente, no volume sanguíneo circulante leva a hipertensão arterial. Se o paciente já sofreu de hipertensão, o nível de pressão arterial aumenta e a terapia anti-hipertensiva torna-se ineficaz. As manifestações estão piorando doença cardíaca coração, aterosclerose cerebral. Como o MCE aumenta gradualmente, a pletora, um aumento no número de glóbulos vermelhos e hemoglobina, sinais de distúrbio da microcirculação em vários pacientes aparecem 2 a 4 anos antes do diagnóstico ser feito.
II – eritrêmico expandido (10-15 anos):
A. Sem metaplasia mieloide do baço
estado geral é perturbado
abundância pronunciada (Hb 200 g/l ou mais)
complicações trombóticas (acidente vascular cerebral, infarto do miocárdio, necrose das pontas dos dedos)
panmielose
eritromelalgia (dor nos membros e ossos)
No quadro do sangue periférico, além da eritrocitose, a neutrofilia costuma estar presente com desvio da fórmula leucocitária para a esquerda para mielócitos únicos, além de basofilia e trombocitose. Na medula óssea, é detectada hiperplasia total de três linhas com megacariocitose pronunciada e é possível mielofibrose reticulina. Mas nesta fase da doença ainda não há metaplasia mieloide do baço (MMS), e a esplenomegalia observada é devida ao aumento do sequestro de eritrócitos e plaquetas. As complicações vasculares são mais frequentes e graves do que na primeira fase da doença. Na patogênese da trombose, um papel importante é desempenhado pelo aumento do MCE, levando ao aumento da viscosidade sanguínea e à desaceleração do fluxo sanguíneo, trombocitose, bem como disfunção do endotélio. A isquemia associada ao fluxo sanguíneo arterial prejudicado é observada em 24-43% dos pacientes. Predomina a trombose de vasos cerebrais, coronárias e artérias que fornecem sangue aos órgãos abdominais. A trombose venosa é detectada em 25-30% dos pacientes e é a causa de morte em aproximadamente um terço dos pacientes que sofrem de PV. A trombose das veias do sistema porta e das veias mesentéricas não é incomum. Em vários pacientes, são as complicações trombóticas que se tornam uma manifestação de PI. A policitemia vera pode ser acompanhada por síndrome hemorrágica: sangramentos nasais frequentes e sangramento após extração dentária. A hipocoagulação baseia-se na desaceleração da conversão do fibrinogênio em fibrina, que ocorre proporcionalmente ao aumento do hematócrito, e na violação da retração do coágulo sanguíneo. Erosão e úlceras do estômago e duodeno consideradas complicações viscerais da IP.
B. Com metaplasia mieloide do baço (MMS).
hepatoesplenomegalia
abundância é moderadamente expressa
panmielose
aumento de sangramento
complicações trombóticas
A esplenomegalia aumenta, o número de leucócitos aumenta, o deslocamento da fórmula leucocitária para a esquerda torna-se mais pronunciado. Na medula óssea - panmielose; A mielofibrose de reticulina e colágeno focal se desenvolve gradualmente. O número de glóbulos vermelhos e plaquetas diminui um pouco devido ao aumento da destruição no baço, bem como à substituição gradual do tecido hematopoiético por tecido fibroso. Nesta fase, pode-se observar a estabilização do quadro do paciente, o nível de hemoglobina, glóbulos vermelhos e plaquetas aproxima-se do normal sem medidas terapêuticas.
III – anêmico:
sm anêmico (até mesmo pancitopenia)
mielofibrose grave
fígado, baço aumentado
Na medula óssea, a mielofibrose do colágeno aumenta e a mielopoiese diminui. O hemograma mostra anemia, trombocitopenia e pancitopenia. O quadro clínico da doença pode incluir anemia e síndrome hemorrágica s, esplenomegalia e caquexia aumentam. O resultado da doença pode ser a transformação em leucemia aguda e síndrome mielodisplásica (SMD).
Diagnóstico. Atualmente, os critérios desenvolvidos pelo American Policitemia Vera Study Group (PVSG) são utilizados para estabelecer o diagnóstico de policitemia vera. Você-
1) aumento da massa de glóbulos vermelhos circulantes (mais de 36 ml/kg para homens e mais de 32 ml/kg para mulheres);
2) saturação normal do sangue arterial com oxigênio (pO2 superior a 92%);
3) esplenomegalia.
1) trombocitose (contagem de plaquetas superior a 400 x 10 9 /l);
2) leucocitose (o número de leucócitos é superior a 12 x 10 9 /lb sem sinais de infecção);
3) atividade fosfatase alcalina(neutrófilos acima de 100 unidades na ausência de febre ou infecção);
4) alto teor vitamina B12 (mais de 900 pg/ml).
O diagnóstico de IP é considerado confiável se o paciente apresentar todos os três sinais da categoria A, ou se o primeiro e o segundo sinais da categoria A e quaisquer dois sinais da categoria B estiverem presentes.
Atualmente, o sinal diagnóstico mais importante é o quadro histológico característico da medula óssea; hiperplasia de células de linhagens eritróides, granulocíticas e megacariócitos com predomínio de eritróides, acúmulos de megacariócitos polimórficos (de pequenos a gigantes). A mielofibrose raramente é observada no momento do diagnóstico, mas torna-se distinta com o longo curso da doença.
No estágio I, a policitemia vera, caracterizada por eritrocitose isolada, deve ser diferenciada da eritrocitose secundária, que é uma resposta a qualquer processo patológico do organismo e pode ser verdadeira ou relativa.
A eritrocitose relativa é consequência da hemoconcentração, ou seja, o MCE é normal, mas o volume plasmático está reduzido, o que é observado com desidratação do corpo (por exemplo, uso de diuréticos, poliúria em pacientes com diabetes, vômitos e diarreia), perda de uma grande quantidade de plasma devido a queimaduras.
A verdadeira eritrocitose secundária (MCE está aumentada, o hematócrito está aumentado) é causada pelo aumento da produção de eritropoetina. Este último é de natureza compensatória e é causado pela hipóxia tecidual em pessoas que vivem em altitudes significativas acima do nível do mar, em pacientes com patologias dos sistemas cardiovascular e respiratório e em fumantes. Esta categoria também inclui pacientes com hemoglobinopatias hereditárias, caracterizadas por maior afinidade da hemoglobina pelo oxigênio, do qual uma quantidade menor é liberada nos tecidos do corpo. A produção inadequada de eritropoietina é observada em doenças renais (hidronefrose, patologia vascular, cistos, tumores, anomalias congênitas), câncer hepatocelular, grandes miomas uterinos. Uma característica diagnóstica diferencial essencial é o nível de eritropoetina sérica.
Tratamento. Nos estágios iniciais da doença, recomenda-se a utilização de sangria, o que ameniza significativamente as manifestações da síndrome pletórica. O método de escolha para redução do hematócrito (e da hemoglobina para valores normais) é a sangria (exfusão), recomendada se o hematócrito ultrapassar 0,54. O objetivo do tratamento é um hematócrito inferior a 0,42 para mulheres e 0,45 para homens.Nas condições modernas, a sangria pode ser substituída pela eritrocitoférese. Além disso, para facilitar a sangria e prevenir complicações trombóticas, os pacientes recebem cursos de terapia desagregante (aspirina, reopoliglucina, etc.). A escolha de um método de tratamento no estágio II avançado da PI talvez seja a tarefa mais difícil. Além da eritrocitose, os pacientes apresentam leucocitose e trombocitose, podendo esta última atingir números muito elevados. Alguns pacientes já sofreram algum tipo de complicação trombótica e as exfusões aumentam o risco de trombose.
Ao individualizar a terapia, a idade dos pacientes deve ser levada em consideração. É o tratamento de pacientes com menos de 50 anos, sem histórico de complicações trombóticas e hipertrombocitose grave (< 1000,0 х 10 9 /л) может быть ограничено только кровопусканиями в сочетании с терапией аспирином (или без него) в дозе 100-375 мг в день.
Para pacientes com mais de 70 anos, com história de complicações trombóticas e hipertrombocitose grave, está indicada terapia com drogas mielossupressoras. Pacientes entre 50 e 70 anos de idade sem complicações trombóticas ou hipertrombocitose grave podem ser tratados com agentes mielossupressores ou flebotomia, embora este último tratamento possa aumentar o risco de complicações trombóticas.
Atualmente, além dos agentes sangüíneos e antiplaquetários, a hidroxiureia e o alfa-interferon são usados principalmente para o tratamento da PV, menos frequentemente o busulfan, e a anagrelida é usada no exterior. A hidroxiureia pode ser a droga de escolha em pacientes com PV com leucocitose e trombocitose graves. Mas para os doentes jovem o uso da hidroxiureia é limitado pelos seus efeitos mutagênicos e leucêmicos. Além da hidroxiureia, o interferon-alfa é amplamente utilizado no tratamento da PV. Em primeiro lugar, o IF-a suprime muito bem a proliferação patológica e não tem efeito leucêmico. Em segundo lugar, tal como a hidroxiureia, reduz significativamente a produção de plaquetas e glóbulos brancos. Atenção especial merece a capacidade do IF-a de eliminar a coceira na pele causada pela ingestão procedimentos de água.
Aspirina em dose diária 50-250 mg, via de regra, elimina distúrbios da microcirculação. Tomar este medicamento ou outros agentes antiplaquetários com terapêutica ou para fins preventivos Recomendado para todos os pacientes IP.
Infelizmente, atualmente não existe tratamento eficaz para PV anêmica em estágio III. A terapia é limitada a paliativos. A síndrome anêmica e hemorrágica é corrigida por transfusões de hemocomponentes. Foi relatada a eficácia do transplante de células-tronco hematopoiéticas em pacientes com PV no estágio de mielofibrose com esplenomegalia e pancitopenia e transformação em leucemia aguda ou SMD. A taxa de sobrevivência de três anos dos pacientes após o transplante foi de 64%.
Previsão. Apesar do curso longo e em alguns casos favorável, a PI é doença grave e está repleto de complicações fatais que encurtam a expectativa de vida dos pacientes. Maioria causa comum As mortes dos pacientes são trombose e embolia (30-40%). Em 20-50% dos pacientes no estágio de mielofibrose pós-policitêmica ( Estágio III A transformação IP) ocorre em leucemia aguda, que tem prognóstico desfavorável - a sobrevida em três anos é de apenas 30%.
A policitemia é uma doença que pode ser presumida apenas olhando para o rosto do paciente. E se você fizer também o exame de sangue necessário, não haverá dúvida alguma. Nos livros de referência também pode ser encontrada com outros nomes: eritremia e doença de Vaquez.
A vermelhidão do rosto é bastante comum e sempre há uma explicação para isso. Além disso, é de curto prazo e não dura muito. Vários motivos podem causar vermelhidão repentina rosto: febre, aumento da pressão arterial, queimaduras solares recentes, situação estranha, e pessoas emocionalmente instáveis geralmente tendem a corar com frequência, mesmo que as pessoas ao seu redor não vejam nenhum pré-requisito para isso.
A policitemia é diferente. Aqui A vermelhidão é persistente, não transitória, distribuída uniformemente por todo o rosto. A cor da abundância excessivamente “saudável” é cereja rica e brilhante.
A policitemia vera (eritremia, doença de Vaquez) pertence ao grupo das hemoblastoses (eritrocitose) ou crônicas de curso benigno. A doença é caracterizada pela proliferação dos três germes da hematopoiese com vantagem significativa dos eritrócitos e megacariocíticos, devido aos quais há aumento não só do número de vermelhos células sanguíneas– , mas também o resto das células sanguíneas que se originam desses brotos, onde fonte processo tumoral são as células precursoras da mielopoiese afetadas. São eles que iniciam a proliferação e diferenciação descontrolada em formas maduras de glóbulos vermelhos.

Os que mais sofrem nessas condições são os glóbulos vermelhos imaturos, hipersensíveis à eritropoietina mesmo em pequenas doses. Para policitemia Ao mesmo tempo, observa-se aumento de leucócitos da série granulocítica(principalmente banda e neutrófilos)e plaquetas. As células da série linfóide, que incluem os linfócitos, não são afetadas pelo processo patológico, pois provêm de um germe diferente e possuem um caminho diferente de reprodução e maturação.
Eritremia não quer dizer que ocorra o tempo todo, mas em uma cidade de 25 mil habitantes há algumas pessoas e, por alguma razão, homens com cerca de 60 anos de idade “gostam” mais dessa doença, embora qualquer um possa encontrar essa idade patológica. É verdade, para recém-nascidos e crianças idade mais jovem policitemia vera é absolutamente incomum, portanto se eritremia for detectada em uma criança, então provavelmente ela carregará personagem secundário e ser sintoma e consequência de outra doença (dispepsia tóxica, eritrocitose de estresse).
Para muitas pessoas, a doença classificada como leucemia (e não importa: aguda ou crônica) está principalmente associada ao câncer no sangue. Aqui é interessante descobrir: é câncer ou não? Nesse caso, seria mais conveniente, mais claro e mais correto falar da malignidade ou benignidade da policitemia vera para determinar a fronteira entre o “bem” e o “mal”. Mas, como a palavra “câncer” se refere a tumores de tecidos epiteliais, então em nesse caso este termo é inapropriado porque este tumor vem de tecido hematopoiético.
A doença de Váquez refere-se a Tumores malignos , mas é caracterizado por alta diferenciação celular. O curso da doença é longo e crônico, por enquanto qualificado como benigno. No entanto, tal curso só pode durar até certo ponto, e então, com o direito e tratamento oportuno, mas após algum período de tempo, quando ocorrem alterações significativas na eritropoiese, a doença se transforma em forma aguda e adquire mais características e manifestações “malignas”. É assim que é – a verdadeira policitemia, cujo prognóstico dependerá inteiramente da rapidez com que progride.
 Qualquer paciente que sofre de eritremia, mais cedo ou mais tarde, faz a pergunta: “Por que essa “doença” aconteceu comigo?” Procurando a razão de muitos condições patológicas, via de regra, é útil e dá certos resultados, aumenta a eficácia do tratamento e promove a recuperação. Mas não no caso da policitemia.
Qualquer paciente que sofre de eritremia, mais cedo ou mais tarde, faz a pergunta: “Por que essa “doença” aconteceu comigo?” Procurando a razão de muitos condições patológicas, via de regra, é útil e dá certos resultados, aumenta a eficácia do tratamento e promove a recuperação. Mas não no caso da policitemia.
As causas da doença só podem ser presumidas, mas não declaradas de forma inequívoca. Só pode haver uma pista para um médico descobrir a origem da doença - anomalias genéticas. No entanto, o gene patológico ainda não foi encontrado, portanto a localização exata do defeito ainda não foi determinada. Há, no entanto, sugestões de que a doença de Vaquez possa estar associada à trissomia 8 e 9 pares (47 cromossomos) ou a outro distúrbio do aparelho cromossômico, por exemplo, perda de uma seção (deleção) do braço longo C5, C20, mas estas ainda são suposições, embora baseadas em conclusões de pesquisas científicas.
Se não há nada a dizer sobre as causas da policitemia, muito pode ser dito sobre as manifestações clínicas. São brilhantes e variados, pois já a partir do 2º estágio de desenvolvimento da doença, literalmente todos os órgãos estão envolvidos no processo. As sensações subjetivas do paciente são de natureza geral:
Queixas características desta doença e caracterizadas por ela:
É óbvio que causa todas essas reclamações - distúrbio de microcirculação.

Como desenvolvimento adicional A doença desenvolve cada vez mais novos sintomas:
Devido ao fato de que para policitemia nos primeiros estágios é típico assintomático , as manifestações acima não ocorrem em um dia, mas se acumulam gradativamente e ao longo de um longo período de tempo, no desenvolvimento da doença costuma-se distinguir 3 estágios.
Estado inicial. O estado do paciente é satisfatório, os sintomas são moderados, a duração da fase é de cerca de 5 anos.
Estágio de manifestações clínicas avançadas. Acontece em duas etapas:
II A – ocorre sem metaplasia mieloide do baço, estão presentes sintomas subjetivos e objetivos de eritremia, a duração do período é de 10-15 anos;
II B – aparece metaplasia mieloide do baço. Esta fase é caracterizada por um quadro claro da doença, os sintomas são pronunciados, o fígado e o baço estão significativamente aumentados.
Estágio terminal, apresentando todos os sinais de um processo maligno. As queixas do paciente são variadas, “tudo dói, está tudo errado”. Nessa fase, as células perdem a capacidade de diferenciação, criando um substrato para a leucemia, que substitui a eritremia crônica, ou melhor, transforma-se em leucemia aguda.
A fase terminal é caracterizada por um curso particularmente grave (síndrome hemorrágica, ruptura do baço, processos infecciosos e inflamatórios que não podem ser tratados devido à imunodeficiência profunda). Geralmente logo termina em morte.
Assim, a expectativa de vida para policitemia é de 15 a 20 anos, o que pode não ser ruim, principalmente considerando que a doença pode ocorrer após os 60 anos. Isso significa que há alguma perspectiva de viver até 80 anos. Porém, o prognóstico da doença ainda depende mais do seu desfecho, ou seja, da forma de leucemia que a eritremia se transforma no estágio III (leucemia mieloide crônica, mielofibrose, leucemia aguda).
 O diagnóstico da policitemia vera baseia-se principalmente em dados laboratoriais, medindo os seguintes indicadores:
O diagnóstico da policitemia vera baseia-se principalmente em dados laboratoriais, medindo os seguintes indicadores:
Morfologicamente, os glóbulos vermelhos nem sempre mudam e muitas vezes permanecem normais, mas em alguns casos pode ser observada eritremia anisocitose(glóbulos vermelhos de tamanhos diferentes). A gravidade e o prognóstico da doença com policitemia em um exame de sangue geral são indicados pelas plaquetas (quanto mais, mais grave é o curso da doença);
Além dos parâmetros hematológicos, para estabelecer o diagnóstico de policitemia vera, o paciente é encaminhado para exame ultrassonográfico dos órgãos abdominais (aumento do fígado e baço).
E então o paciente aguarda tratamento no setor de hematologia, onde são determinadas as táticas manifestações clínicas, parâmetros hematológicos e estágio da doença. EM medidas terapêuticas A eritremia geralmente inclui:
O regime de tratamento da eritremia é prescrito pelo médico individualmente para cada caso, portanto nossa tarefa é apenas apresentar brevemente ao leitor os medicamentos utilizados no tratamento da doença de Vaquez.
Um papel significativo no tratamento da policitemia é atribuído ao regime de trabalho (reduzindo atividade física), descanso e nutrição. EM Estado inicial doença, quando os sintomas ainda não se manifestam ou se manifestam de forma fraca, o paciente é encaminhado para a tabela nº 15 (geral), embora com algumas ressalvas. Não se recomenda ao paciente consumir alimentos que melhorem a hematopoiese.(fígado, por exemplo) e sugerir revisão da dieta alimentar, dando preferência aos laticínios e produtos vegetais.
 No segundo estágio da doença, é prescrita ao paciente a tabela nº 6, que corresponde à dieta para gota e limita ou exclui completamente peixes e pratos de carne, legumes e azeda. Após receber alta hospitalar, o paciente deve seguir as recomendações do médico durante a observação ambulatorial ou tratamento.
No segundo estágio da doença, é prescrita ao paciente a tabela nº 6, que corresponde à dieta para gota e limita ou exclui completamente peixes e pratos de carne, legumes e azeda. Após receber alta hospitalar, o paciente deve seguir as recomendações do médico durante a observação ambulatorial ou tratamento.
Pergunta: “É possível tratar remédios populares? sons com a mesma frequência para todas as doenças. A eritremia não é exceção. Porém, como já foi observado, o curso da doença e a expectativa de vida do paciente dependem inteiramente do tratamento oportuno, cujo objetivo é alcançar uma remissão longa e estável e atrasar o terceiro estágio pelo maior tempo possível.
Durante o período de calmaria do processo patológico, o paciente ainda deve lembrar que a doença pode retornar a qualquer momento, por isso deve discutir sua vida sem agravamento com o médico assistente com quem está sendo observado, fazer exames e realizar exames periodicamente .
O tratamento de doenças do sangue com remédios populares não deve ser generalizado, e se existem muitas receitas para aumentar ou diminuir os níveis de hemoglobina, isso não significa de forma alguma que sejam adequados para o tratamento da policitemia, da qual, em geral, Ervas medicinais ainda não encontrado. A doença de Vaquez é um assunto delicado e para controlar a função da medula óssea e assim influenciar o sistema hematopoiético é necessário ter dados objetivos que possam ser avaliados por uma pessoa com certo conhecimento, ou seja, o médico assistente.
Concluindo, gostaria de dizer algumas palavras aos leitores sobre a eritremia relativa, que não se confunde com a verdadeira, pois a eritrocitose relativa pode ocorrer no contexto de muitas doenças somáticas e terminar com sucesso quando a doença estiver curada. Além disso, a eritrocitose como sintoma pode acompanhar vômitos prolongados, diarréia, queimaduras e hiperidrose. EM casos semelhantes a eritrocitose é um fenômeno temporário e está associada principalmente à desidratação do corpo, quando a quantidade de plasma circulante, que consiste em 90% de água, diminui.
Policitemia vera (eritremia, doença de Vaquez) refere-se a doenças mieoproliferativas, durante as quais, quando uma célula-tronco é danificada, ocorre a proliferação de seus três brotos. Há um aumento na formação de glóbulos vermelhos e, em menor grau, de leucócitos e plaquetas. Quase sempre com esta doença, é observada metaplasia mieloide do baço.
O número de casos de eritremia é de 1 por 100 mil habitantes, e essa patologia ocorre com mais frequência em homens do que em mulheres. A doença de Vaquez é mais comum entre pessoas com mais de 60 anos de idade do que em outras faixas etárias.
A base da doença é a transformação de uma célula-tronco hematopoiética, o que dá direito a considerar a doença de Vaquez como uma doença neoplásica clonal.
Células pluripotentes sofrem transformação maligna célula tronco, o que leva ao envolvimento de todas as três hematopoiese no processo. As células da linhagem eritróide alcançam a maior proliferação. Como isso ocorre na ausência de eritropoetina, esse sinal é específico da eritremia.
Uma característica da doença de Vaquez também é o acúmulo de megacariócitos polimórficos.
Com o aumento da massa de glóbulos vermelhos circulantes, o hematócrito aumenta e o sangue fica mais viscoso. A combinação desses fatores juntamente com a trombocitose leva à microcirculação prejudicada e complicações tromboembólicas frequentes. Paralelamente, desenvolve-se metaplasia mieloide do baço.
A doença ocorre em 4 estágios, refletindo processos patológicos que ocorrem na medula óssea e no baço.
Na primeira fase da doença observa-se eritrocitose e panmielose na medula óssea. A durabilidade deste aço é de até 5 anos.
A doença nesta fase é acompanhada por pletora, acrocianose, dores ardentes e parestesia nas pontas dos dedos. Alguns pacientes relatam a aparência coceira na pele após a lavagem. O aumento do volume de hemácias circulantes leva ao aparecimento de hipertensão arterial em pacientes que não apresentavam queixa desse sintoma antes do início da doença, ou agravamento de um já existente. hipertensão, que é difícil de tratar com medicamentos anti-hipertensivos tradicionais. Os sintomas de doença coronariana e aterosclerose cerebral tornam-se mais pronunciados.
Como a doença se desenvolve gradualmente, leva de 2 a 4 anos desde o seu início até o diagnóstico.
Segundo estágio A da doença de Vaquez chamado eritrêmico e dura de 10 a 15 anos. Além da eritrocitose, observa-se aumento do conteúdo de neutrófilos com desvio para a esquerda. O quadro da medula óssea nesta fase da doença é caracterizado por hiperplasia total de três linhas com megacariocitose pronunciada. A metaplasia mieloide do baço ainda está ausente, mas há esplenomegalia causada pelo aumento do sequestro de glóbulos vermelhos e plaquetas.
Esta fase da doença é acompanhada por sintomas mais graves e mais frequentes complicações vasculares. O risco de desenvolver complicações trombóticas é maior em pacientes idosos com história de trombose prévia.
Os vasos sanguíneos mais frequentemente trombosados no cérebro são artérias coronárias e vasos que irrigam os órgãos abdominais.
Muitas vezes com policitemia verdadeira, apesar quantidade aumentada plaquetas, observa-se síndrome hemorrágica, manifestada por sangramento nasal e grande perda de sangue após a extração do dente. Isso ocorre devido à lenta conversão de fibrinogênio em fibrina e à retração prejudicada do coágulo sanguíneo.
As complicações viscerais da doença de Vaquez podem incluir erosões e úlceras do estômago e duodeno. São consequência do fluxo sanguíneo prejudicado e do trofismo da mucosa gástrica e intestinal.
Segundo estágio B da eritremia difere pela adição de metaplasia mieloide do baço. A esplenomegalia torna-se mais pronunciada. No sangue periférico observa-se leucocitose, ocorre um deslocamento da fórmula para a esquerda nas formas juvenis. A medula óssea é caracterizada por panmielose. Alguma estabilização da condição dos pacientes nesta fase da doença pode ser devida a uma diminuição no nível de glóbulos vermelhos devido ao aumento da sua destruição pelo baço.
Estágio três da doença de Vaquez foi chamado de anêmico. Além do fenômeno da anemia, são observadas trombocitopenia e pancitopenia no sangue periférico. Paralelamente, aumentam a esplenomegalia e a caquexia.
Critérios que permitem diagnosticar policitemia vera, são divididos em principais e adicionais.
O primeiro inclui um aumento na massa de glóbulos vermelhos circulantes com saturação normal Sangue arterial oxigênio, bem como esplenomegalia.
Os sintomas adicionais incluem trombocitose e leucocitose na ausência de sinais de infecção, aumento da atividade da fosfatase alcalina acima de 100 unidades e níveis elevados de vitamina B12.
Atualmente grande importância No diagnóstico de eritremia, o quadro da medula óssea é anexado.
Para prevenir a trombose, a aspirina é prescrita na dose de 50 a 250 mg por dia.
Muitas vezes, com a correção da microcirculação, os sintomas de coceira após um banho quente também desaparecem. Porém, em alguns casos, para eliminar este sintoma é necessário recorrer à irradiação ultravioleta de sangue autólogo.
Para reduzir o número de glóbulos vermelhos, utiliza-se a sangria, que deve ser feita com extrema cautela. A sangria frequente pode levar à deficiência de ferro, que não requer correção sem sinais de deficiência tecidual desse elemento.
Eritremia grave requer uso métodos medicinais diminuição do número de glóbulos vermelhos. Para tanto, são utilizados medicamentos hidroxiureia, alfa-interferon e mielosan.
Apesar do fato de a doença durar muito tempo e ser relativamente benigna, resultados fatais com a doença de Vaquez são possíveis devido a desenvolvendo complicações doenças. Na maioria das vezes, a vida do paciente é ameaçada por trombose e tromboembolismo, bem como pela transformação da doença em leucemia aguda.
As medidas preventivas para eritremia visam detecção precoce doenças e prevenindo o desenvolvimento de complicações da doença.
A policitemia vera (eritremia, doença de Vaquez ou policitemia primária) é uma leucemia progressiva. doença maligna, que está associada à hiperplasia de elementos celulares da medula óssea (mieloproliferação). O processo patológico afeta principalmente o germe eritroblástico, de modo que um número excessivo de glóbulos vermelhos é detectado no sangue. Há também um aumento no número leucócitos neutrófilos e plaquetas.
| CID-10 | D45 |
|---|---|
| CID-9 | 238.4 |
| CID-O | M9950/3 |
| Medline Plus | 000589 |
| Malha | D011087 |
Um número aumentado de glóbulos vermelhos aumenta a viscosidade do sangue, aumenta a sua massa, provoca uma desaceleração do fluxo sanguíneo nos vasos e a formação de coágulos sanguíneos. Como resultado, os pacientes desenvolvem suprimento sanguíneo prejudicado e hipóxia.
A policitemia vera foi descrita pela primeira vez em 1892 pelo médico e cardiologista francês Vaquez. Vaquez sugeriu que a hepatoesplenomegalia e a eritrocitose detectadas em seu paciente surgiram como resultado do aumento da proliferação de células hematopoiéticas e identificou a eritremia como uma forma nosológica separada.
Em 1903, W. Osler usou o termo “doença de Vaquez” para descrever pacientes com esplenomegalia (baço aumentado) e eritrocitose grave e deu uma descrição detalhada da doença.
Turk (W. Turk) em 1902-1904 sugeriu que nesta doença o distúrbio da hematopoiese é de natureza hiperplásica e chamou a doença de eritremia por analogia com a leucemia.
A natureza neoplásica clonal da mieloproliferação, observada na policitemia, foi comprovada em 1980 por P. J. Fialkov. Ele descobriu um tipo de enzima, a glicose-6-fosfato desidrogenase, em glóbulos vermelhos, granulócitos e plaquetas. Além disso, ambos os tipos desta enzima foram detectados nos linfócitos de dois pacientes heterozigotos para esta enzima. Graças à pesquisa de Fialkov, ficou claro que o alvo do processo neoplásico é a célula precursora da mielopoiese.
Em 1980, vários pesquisadores conseguiram separar o clone neoplásico das células normais. Foi provado experimentalmente que na policitemia se forma uma população de precursores comprometidos com eritróides que apresentam patologicamente alta sensibilidade mesmo a uma pequena quantidade de eritropoietina (hormônio renal). Segundo os cientistas, isso contribui para o aumento da formação de glóbulos vermelhos na policitemia vera.
Em 1981, L. D. Sidorova e coautores realizaram estudos que permitiram detectar alterações qualitativas e quantitativas no componente plaquetário da hemostasia, que desempenham um papel importante no desenvolvimento de complicações hemorrágicas e trombóticas na policitemia.
A policitemia vera é detectada principalmente em idosos, mas pode ser observada em jovens e crianças. Nos jovens, a doença é mais grave. A idade média dos pacientes varia de 50 a 70 anos. A idade média dos que adoecem pela primeira vez aumenta gradualmente (em 1912 era de 44 anos e em 1964 era de 60 anos). O número de pacientes com menos de 40 anos é de cerca de 5%, e a eritremia em crianças e pacientes com menos de 20 anos é detectada em 0,1% de todos os casos da doença.
A eritremia é ligeiramente menos comum em mulheres do que em homens (1: 1,2-1,5).
É a doença mais comum no grupo das doenças mieloproliferativas crônicas. É bastante raro - segundo várias fontes, de 5 a 29 casos por 100.000 habitantes.
Existem dados dispersos sobre a influência de fatores raciais (acima da média entre os judeus e abaixo da média entre os representantes). Raça negróide), no entanto, no momento esta suposição não foi confirmada.
A policitemia vera é dividida em:
Observa-se aumento absoluto da massa eritrocitária em todos os pacientes, mas apenas em 2/3 o número de leucócitos e plaquetas também aumenta.
As causas da policitemia vera não foram definitivamente estabelecidas. Não existe atualmente teoria unificada, o que explicaria a ocorrência de hemoblastoses (tumores sanguíneos), às quais esta doença pertence.
Com base em observações epidemiológicas, foi apresentada uma teoria sobre a ligação da eritremia com a transformação das células-tronco, que ocorre sob a influência de mutações genéticas.
Foi estabelecido que a maioria dos pacientes apresenta uma mutação na enzima Janus quinase-tirosina quinase, sintetizada no fígado, que está envolvida na transcrição de certos genes por meio da fosforilação de muitas tirosinas na parte citoplasmática dos receptores.
A mutação mais comum, descoberta em 2005, está no éxon 14 JAK2V617F (detectada em 96% de todos os casos da doença). Em 2% dos casos, a mutação afeta o exon 12 do gene JAK2.
Pacientes com policitemia vera também apresentam:
Pacientes idosos com alta carga alélica JAK2V617F são caracterizados por nível aumentado hemoglobina, leucocitose e trombocitopenia.
Com uma mutação do gene JAK2 no éxon 12, a eritremia é acompanhada por um nível sérico subnormal do hormônio eritropoietina. Os pacientes com esta mutação são mais jovens.
Na policitemia vera, mutações de TET2, IDH, ASXL1, DNMT3A, etc. também são frequentemente detectadas, mas são significado patogenético ainda não foi estudado.
Diferenças na sobrevida de pacientes com tipos diferentes nenhuma mutação foi detectada.
Como resultado de distúrbios genéticos moleculares, a via de sinalização JAK-STAT é ativada, que se manifesta pela proliferação (produção celular) da linhagem mieloide. Ao mesmo tempo, aumenta a proliferação e o aumento do número de glóbulos vermelhos no sangue periférico (também é possível um aumento no número de leucócitos e plaquetas).
As mutações identificadas são herdadas de forma autossômica recessiva.
Existe também a hipótese segundo a qual a causa da eritremia pode ser vírus (foram identificados 15 tipos desses vírus), que, na presença de fatores predisponentes e imunidade enfraquecida, penetram nas células imaturas da medula óssea ou nos gânglios linfáticos. As células afetadas pelo vírus começam a se dividir ativamente em vez de amadurecer, iniciando assim o processo patológico.
Os fatores que provocam a doença incluem:
A eritremia secundária se desenvolve sob a influência de fatores favoráveis quando:
A patogênese da policitemia vera está associada a uma violação do processo de hematopoiese (hematopoiese) ao nível da célula progenitora. A hematopoiese adquire a proliferação ilimitada de células progenitoras características de um tumor, cujos descendentes formam um fenótipo especializado em todas as linhagens hematopoiéticas.
A policitemia vera é caracterizada pela formação de colônias eritróides na ausência de eritropoietina exógena (o aparecimento de colônias endógenas independentes de eritropoetina é um sinal que distingue a eritremia da eritrocitose secundária).
A formação de colônias eritróides indica uma interrupção na implementação dos sinais regulatórios que a célula mieloide recebe do ambiente externo.
A base da patogênese da policitemia vera são defeitos nos genes que codificam proteínas responsáveis por manter a mielopoiese dentro da faixa normal.
Uma diminuição na concentração de oxigênio no sangue causa uma reação nas células intersticiais dos rins que sintetizam a eritropoetina. O processo que ocorre nas células intersticiais diz respeito ao trabalho de muitos genes. A principal regulação desse processo é realizada pelo fator 1 (HIF-1), que é uma proteína heterodimérica composta por duas subunidades (HIF-1alfa e HIF-1beta).
Se a concentração de oxigênio no sangue estiver dentro dos limites normais, os resíduos de prolina (o aminoácido heterocíclico da molécula HIF-1 existente livremente) são hidroxilados sob a influência da enzima reguladora PHD2 (sensor de oxigênio molecular). Graças à hidroxilação, a subunidade HIF-1 adquire a capacidade de se ligar à proteína VHL, o que proporciona prevenção tumoral.
A proteína VHL forma um complexo com uma série de proteínas E3 ubiquitina ligase, que, após formar ligações covalentes com outras proteínas, são enviadas ao proteassoma e ali destruídas.
Durante a hipóxia, a hidroxilação da molécula HIF-1 não ocorre; as subunidades desta proteína se combinam e formam a proteína heterodimérica HIF-1, que viaja do citoplasma para o núcleo. Uma vez no núcleo, a proteína liga-se a sequências especiais de DNA nas regiões promotoras dos genes (a conversão dos genes em proteína ou RNA é induzida pela hipóxia). Como resultado dessas transformações, a eritropoietina é liberada na corrente sanguínea pelas células intersticiais dos rins.
Nas células precursoras da mielopoiese, o programa genético incorporado nelas é realizado como resultado do efeito estimulante das citocinas (essas pequenas moléculas peptídicas de controle (sinal) ligam-se aos receptores correspondentes na superfície das células precursoras).
Quando a eritropoietina se liga ao receptor de eritropoietina EPO-R, ocorre a dimerização desse receptor, que ativa Jak2, uma quinase associada aos domínios intracelulares do EPO-R.
A Jak2 quinase é responsável pela transmissão do sinal da eritropoietina, trombopoietina e G-CSF (fator estimulador de colônias de granulócitos).
Devido à ativação da Jak2-quinase, ocorre a fosfolação de uma série de proteínas alvo citoplasmáticas, que incluem proteínas adaptadoras da família STAT.
A eritremia foi detectada em 30% dos pacientes com ativação constitutiva do gene STAT3.
Além disso, na eritremia, em alguns casos, é detectado um nível reduzido de expressão do receptor de trombopoietina MPL, que é de natureza compensatória. A redução na expressão de MPL é secundária e causada por um defeito genético responsável pelo desenvolvimento da policitemia vera.
Uma diminuição na degradação e um aumento no nível do fator HIF-1 são causados por defeitos no gene VHL (por exemplo, representantes da população da Chuváchia são caracterizados por uma mutação homozigótica 598C>T deste gene).
A policitemia vera pode ser causada por anomalias no cromossomo 9, mas a mais comum é a deleção do braço longo do cromossomo 20.
Em 2005, foi identificada uma mutação pontual no éxon 14 do gene Jak2 quinase (mutação JAK2V617F), que provoca a substituição do aminoácido valina por fenilalanina no domínio pseudoquinase JH2 da proteína JAK2 na posição 617.
A mutação JAK2V617F em células precursoras hematopoiéticas na eritremia apresenta-se na forma homozigótica (a formação da forma homozigótica é afetada pela recombinação mitótica e duplicação do alelo mutante).
Quando JAK2V617F e STAT5 estão ativos, o nível de espécies reativas de oxigênio aumenta, resultando em uma transição do ciclo celular da fase G1 para a fase S. Proteína adaptadora STAT5 e formulários ativos o oxigênio transmite um sinal regulatório de JAK2V617F para os genes ciclina D2 e p27kip, o que causa uma transição acelerada do ciclo celular da fase G1 para a fase S. Como resultado, a proliferação de células eritróides que carregam uma forma mutante do gene JAK2 aumenta .
Em pacientes positivos para JAK2V617F, esta mutação é detectada em células mieloides, linfócitos B e T e células natural killer, o que comprova a vantagem proliferativa das células defeituosas em comparação com a norma.
A policitemia vera, na maioria dos casos, é caracterizada por uma proporção bastante baixa de alelo mutante para normal em células mieloides maduras e precursores iniciais. Na presença de dominância clonal, os pacientes apresentam quadro clínico em comparação com pacientes sem esse defeito.
Os sintomas da policitemia vera estão associados à produção excessiva de glóbulos vermelhos, que aumentam a viscosidade do sangue. Na maioria dos pacientes, o nível de plaquetas, que causa trombose vascular, também aumenta.
A doença se desenvolve muito lentamente e Estado inicialé assintomático.
Para mais fases posteriores A policitemia vera se manifesta:
A síndrome pletórica é acompanhada por:
A síndrome mieloproliferativa se manifesta:
Também são observadas varizes, especialmente visíveis na região do pescoço, sinal de Cooperman (mudança na cor do palato mole com coloração normal do palato duro), úlcera duodenal e, em alguns casos, estômago, sangramento de gengiva e esôfago, e aumento dos níveis de ácido úrico. É possível o desenvolvimento de insuficiência cardíaca e cardiosclerose.
A policitemia vera é caracterizada por três estágios de desenvolvimento:
A eritremia é diagnosticada com base em:
Também são realizados um coagulograma, estudos do metabolismo do ferro e determinado o nível de eritropoietina no soro sanguíneo.
Como a eritremia crônica é acompanhada por aumento do fígado e do baço, é realizada uma ultrassonografia dos órgãos internos. O ultrassom também detecta a presença de hemorragias.
Para avaliar a extensão do processo tumoral, SCT (espiral Tomografia computadorizada) e ressonância magnética (ressonância magnética).
Para identificar anormalidades genéticas, é realizado um estudo genético molecular do sangue periférico.
Os objetivos do tratamento da policitemia vera são:
A eritremia é tratada com:
Para prevenir a trombose, são utilizados anticoagulantes (geralmente é prescrito ácido acetilsalicílico na dose de 40-325 mg/dia).
A nutrição para eritremia deve atender aos requisitos da tabela de tratamento de acordo com Pevzner nº 6 (reduz-se a quantidade de alimentos protéicos, excluem-se frutas e vegetais vermelhos e alimentos que contenham corantes).

Em 2018, o Jejum da Natividade começará em 28 de novembro. Durante este período, os crentes ortodoxos se preparam para celebrar o Natal...

Constituir família é o sonho da maioria das mulheres. Eles querem ter um marido amoroso e um monte de filhos. Mas nem sempre é um relacionamento...

Este artigo contém: a oração mais poderosa pelo divórcio - informações retiradas de todo o mundo, da rede eletrônica e...

Site de informações sobre ícones, orações, tradições ortodoxas. Oração pelos escândalos e brigas na família, com o marido, com os filhos...

O que seria do Ano Novo sem champanhe, tangerinas, Olivier, formol e o “arenque com casaco de pele” preferido de todos. Com o último...

Vamos preparar os ingredientes necessários para os biscoitos. A primeira coisa a fazer é colocar a água para ferver. Nós precisamos...

É possível cadastrar funcionário para o cargo de diretor financeiro - contador-chefe? O contador-chefe afirma...

O chefe de uma pequena empresa pode gerenciar facilmente o orçamento de forma independente. VERIFICADO! Se você conseguir...

A criação de novos projetos envolve uma justificação económica preliminar para a sua viabilidade, posterior...
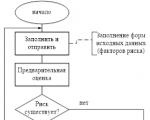
O reporte é gerado pelo RM, é acordado (aprovado) pelo Comité de Risco no âmbito do Conselho de Administração e transmitido a...

Na beira de um prado grande, muito grande, em uma longa folha de grama esmeralda vivia uma pequena joaninha. O pequeno de Deus...

Hoje em dia é bastante comum as pessoas se voltarem para as estrelas. Com a ajuda de um horóscopo uma pessoa pode descobrir...

Negócios ou amigável. Se você estava perseguindo um estranho, isso significa o seu nível de confiança no sexo feminino...

de acordo com o livro dos sonhos de Freud Se você sonhou que estava pescando, isso significa que na vida real dificilmente você consegue desligar...

Constituir família é o sonho da maioria das mulheres. Eles querem ter um marido amoroso e um monte de filhos. Mas nem sempre é...

Este artigo contém: a oração mais poderosa pelo divórcio - informações retiradas de todo o mundo, eletrônicas...