Какво представлява дуалността вълна-частица?
Двойствеността вълна-частица на светлината означава, че светлината едновременно има свойствата на непрекъснато електромагнитно...
Колкото и невероятно да изглежда, прогерията всъщност задейства механизми за преждевременно стареене в младия организъм. Официално заболяването е кръстено на учените, които първи са описали и изследвали патологията: при деца това е синдром на Хътчинсън-Гилфорд, при възрастни - синдром на Вернер.
Прогерията се среща няколко пъти по-често при момчетата, отколкото при момичетата. Средно пациентите живеят от 10 до 13 години (в изключителни случаидо 20): смъртоносна болест, за съжаление, не дава шанс за възстановяване и дълги годиниживот. Такива деца значително изостават физическо развитиеот здрави връстници, но това не са всички „прелести“ на прогерията. Силно изтощениетяло, нарушение на структурата на кожата, липса на вторични знациполовото развитие и косата, недоразвити вътрешни органии външният вид на стареца като цяло - това е бремето, което пада върху плещите на нещастното дете.
IN умствено развитиедетето е абсолютно адекватно, тялото му запазва детски пропорции, но в същото време епифизният хрущял бързо надрасна и на негово място се появява епифизна линия - всичко е като при възрастен. Бързо растящите деца са принудени да се изправят далеч от детските проблеми, свързани с прогерията: атеросклероза, инсулт и различни сърдечни заболявания.

За съжаление, експертите все още не са разгледали в детайли истинското лице на „врага“. В резултат на продължителни изследвания учените успяха да разберат, че в основата на патологията най-вероятно е мутация на ламиновия ген (LMNA), който е пряко свързан с процеса на клетъчно делене. Срив в генетичната система лишава клетките от устойчивост и задейства неочаквани механизми на стареене в тялото.
Прогерията, за разлика от много други заболявания с генетична етиология, не се предава по наследство, тоест се появява напълно случайно и никой от родителите на болно дете не може да се нарече носител на патологията.

Непосредствено след раждането децата, носители на смъртоносния ген на прогерията, не могат да бъдат разграничени от здрави бебета. Още през първата година от живота многобройните симптоми на заболяването се усещат напълно. Между тях:
След като дете, страдащо от прогерия, празнува петия си рожден ден, тялото му започва неумолим процес на развитие, при който са особено засегнати аортата, мезентериалните и коронарните артерии. На фона на тези нарушения се отбелязва появата на сърдечни шумове и хипертрофия на лявата камера. Комплексният ефект на тези нарушения върху тялото се счита за една от причините за краткия живот на пациентите с прогерия. Основният фактор за внезапната смърт на пациентите се нарича още исхемичен инсулт.

Болестта може внезапно да изпревари възрастен на възраст от 14-15 до 18 години. Пациентът започва да губи тегло, да закърнява, да побелява и постепенно да оплешивява (прогресивна алопеция). Кожата на човек с прогерия става по-тънка, губи всичките си цветове, придобивайки нездравословен блед нюанс. Отдолу ясно се вижда мрежа от кръвоносни съдове, подкожната мастна тъкан и мускулите напълно атрофират, така че ръцете и краката изглеждат много тънки.
При пациенти над 30 години и двете очни ябълкизасяга, гласът отслабва, кожата над издатините на костите става груба и покрита с язви. Страдащите от прогерия изглеждат по същия начин: нисък ръст, лице с форма на луна, нос, наподобяващ птичи клюн, тясна уста, рязко изпъкнала брадичка, плътно тяло и тънки сухи крайници, обезобразени от множество пигментни петна. Болестта безцеремонно пречи на различни системи на тялото: работата на потта и мастни жлези, нормалната дейност на сърдечно-съдовата система е нарушена, тялото страда от калцификация, остеопороза и ерозивен остеоартрит. За разлика от малките пациенти, при възрастните заболяването оказва пагубно влияние върху интелектуални способности.
Около 10% от пациентите до четиридесетгодишна възраст са изправени пред такива ужасни заболявания като саркома, астроцитом и меланом. Онкологията се развива на фона на захарен диабет и нарушени функции паращитовидни жлези. Непосредствена причинасмъртните случаи на пациенти с прогерия в повечето случаи са злокачествени тумории сърдечно-съдови патологии.
Външните симптоматични прояви на патологията са толкова ярки и красноречиви, че заболяването се диагностицира въз основа на клиничната картина.

МирСоветов е принуден да признае, че за съжаление няма панацея за прогерия. Всички методи на лечение, които се използват днес, също не винаги са ефективни. Лекарите обаче правят всичко, което зависи от тях. Така че всички пациенти са под ред медицинско наблюдение, тъй като чрез наблюдение на състоянието на сърдечно-съдовата система е възможно своевременно да се открие развитието на усложнения на определено „сърдечно“ заболяване.
Всички методи на лечение преследват една единствена, но жизненоважна цел - да „замразят“ болестта, да не позволят да се влоши и да облекчат състоянието на пациента, доколкото е възможно съвременна медицина. Как могат да помогнат специалистите?
Превенцията на заболяването все още не е разработена.
Какво е прогерия, какви са нейните признаци и последствия? Как се диагностицира заболяването и какво лечение се предлага днес?
 Синдромът на Hutchinson-Gilford Progeria (HGS) е рядко, фатално генетично заболяване, характеризиращо се с внезапно, ускорено стареене при децата и засяга едно на всеки 8 милиона деца по света. Името на синдрома идва от гръцката дума и означава „преждевременно стареене“. Въпреки че има различни формипрогерия, класически тип синдром на Хътчинсън-Гилфорд Прогерия е кръстена на лекарите, които за първи път описват болестта, през 1886 г. от д-р Джонатан Хътчинсън и през 1897 г. от д-р Хейстингс Гилфорд.
Синдромът на Hutchinson-Gilford Progeria (HGS) е рядко, фатално генетично заболяване, характеризиращо се с внезапно, ускорено стареене при децата и засяга едно на всеки 8 милиона деца по света. Името на синдрома идва от гръцката дума и означава „преждевременно стареене“. Въпреки че има различни формипрогерия, класически тип синдром на Хътчинсън-Гилфорд Прогерия е кръстена на лекарите, които за първи път описват болестта, през 1886 г. от д-р Джонатан Хътчинсън и през 1897 г. от д-р Хейстингс Гилфорд.
Днес е известно, че CSGP се причинява от мутация в гена LMNA (Lamin). Генът LMNA произвежда протеина ламин, който държи клетъчното ядро заедно. Изследователите смятат, че дефектен протеин ламин прави клетъчните ядра нестабилни. И именно тази нестабилност стартира процеса преждевременно стареене.
Децата с този синдром изглеждат здрави при раждането, първите физически признаци на заболяването могат да се появят на възраст от една и половина до две години. Това спиране на растежа, загуба на тегло и коса, изпъкнали вени, набръчкана кожа - всичко това е съпроводено с усложнения, по-типични за възрастните хора - скованост на ставите, генерализирана атеросклероза, остеопороза, сърдечно-съдови заболявания, инсулт. Децата с това състояние имат забележително подобен външен вид, въпреки различния си етнически произход. Най-често децата с прогерия умират от (сърдечно заболяване) на средна възраст от тринадесет години (диапазон от около 8 до 21 години).
Има и "възрастна" прогерия (синдром на Вернер), която започва в юношеството(15-20 години). Продължителността на живота на пациентите се намалява до 40-50 години. Повечето често срещани причини фатален изходса миокарден инфаркт, инсулт и злокачествени тумори. Учените не могат да определят точната причина за заболяването.
Въпреки че прогерията е генетично заболяване, в класическия смисъл на синдрома на Hutchinson-Gilford, то не е наследствено, т.е. Никой от родителите не е носител или засегнат. Смята се, че всеки случай представлява спорадична (случайна) мутация, която се появява или в яйцеклетката, или в спермата преди зачеването.
Заболяването засяга всички раси и двата пола по равно. Ако двойка родители имат едно дете с CSGP, шансът второ дете да се роди със същото заболяване е 1 на 4 до 8 милиона. Има други прогерични синдроми, които могат да се предават от поколение на поколение, но не и класическият CSGP.
След като тази генна мутация е идентифицирана, Progeria Research Foundation разработи програми за диагностични тестове. Вече е възможно да се потвърдят специфичните генетични промени или мутации в ген, които водят до CSGP. След първоначална клинична оценка (външен вид на детето и медицинска документация), на детето се взема кръвна проба за изследване. В момента се разработва окончателен научен метод за диагностициране на деца. Това ще доведе до по-точни и др ранна диагностика, което ще помогне да се гарантира, че децата с тази мутация получават подходящи грижи.
На пръв поглед обикновена форма психологическо състояние- фобична тревожност, причинява увреждане на клетките и води до преждевременно стареене.
Към днешна дата има само няколко възможности за оптимизиране на качеството на живот на децата с прогерия. Лечението включва продължителни грижи, сърдечни грижи, специална хранаи физическа терапия.
През последните няколко години бяха публикувани обнадеждаващи данни от изследвания, описващи потенциала лечение с лекарстваза деца с прогерия. Учените вярват, че инхибиторите на фарнезилтрансферазата (FTI), първоначално разработени за лечение на рак, могат да обърнат структурните аномалии, които причиняват прогерия при деца.
26 деца са участвали в опити за наркотици - това е една трета от всички известни случаипрогерия. Децата, приемащи лекарството, показват 50% увеличение на годишното наддаване на тегло. Плътността също се подобрява при децата костна тъканпреди нормално ниво, и 35% намаление на артериалната скованост, което се свързва с висок рисксърдечен удар. Изследователите подчертават, че благодарение на новата разработка увреждането на кръвоносните съдове не само намалява, но и частично се възстановява за периода.
Всички хора остаряват. Според учените, ако не за разрушително въздействие външна средаи нашите разрушителни пристрастявания към удоволствия, вредни за тялото, бихме живели до 130 или дори 150 години. А преди 16 години, на 29 август 2001 г., учените дори обявиха, че са открили ген на дълголетието. Така че може би в близко бъдеще ще можем да изживеем целия живот, определен ни от природата. Но засега ние остаряваме и умираме, по-голямата част преди 80-90-годишна възраст. А някои заболявания скъсяват този и без това не толкова дълъг период няколко пъти. И най-„убиецът“ сред тях, в буквалния смисъл на думата, е прогерията. Разбрахме какво е да остарееш след едно и половина до две десетилетия.
Стареенето е естествен процес, характерен за всеки жив организъм на Земята. Всички съществуващи теории по темата „Защо хората остаряват?“ може да се раздели на две големи групи. Поддръжниците на един от тях твърдят, че стареенето е предназначено от природата за по-нататъшното развитие на видовете и обществото. Други са сигурни, че тук няма глобални планове - просто щетите на генно и клетъчно ниво се натрупват с течение на времето, което води до износване на тялото.
По един или друг начин, но наистина в хода на живота на човека резултатите от вътрешни неуспехи и грешки се натрупват в неговите клетки и тъкани, както и последствията външни влияния. Между ключови фактористареене са следните:
Прогерията, за която ще стане дума в тази статия, не е стареене - в смисъла, в който науката я разбира, когато говори за продължителност на живота, износване на тялото и т.н. Това заболяване изглежда като стареене, въпреки че всъщност е тежко генетично заболяване, свързано с нарушено производство на определени протеини.
Англичанинът Джонатан Хътчинсън през 1886 г. за първи път описва 6-годишно дете, при което наблюдава атрофия на кожата. А името на необичайното заболяване (от гръцката дума „progeros” – старо преди времето си) му е дадено през 1897 г. от д-р Гилфорд, който изучава и описва нюансите на заболяването. И през 1904 г. д-р Вернер публикува описание на прогерия при възрастни - като използва примера на четирима братя и сестри, които страдат от катаракта и склеродермия.
Смята се, че Франсис Скот Фицджералд е написал своя разказ „Странният случай на Бенджамин Бътън“ през 1922 г. именно под впечатлението от информация за пациенти с прогерия. През 2008 г. Брад Пит изигра главния герой на книгата във филма Мистериозна историяБенджамин Бътън."
Има два вида прогерия:
Това е рядка патология. Среща се при 1 дете от няколко милиона. Смята се, че днес в света има не повече от сто души, страдащи от детска прогерия. Учените обаче предполагат, че можем да говорим за още около 150 недиагностицирани случая.
Също така е рядко заболяване, но не толкова, колкото детската прогерия. Хората със синдром на Вернер се раждат в 1 случай на 100 хиляди. В Япония - малко по-често: 1 случай на 20-40 хиляди новородени. Общо в света са известни малко по-малко от 1,5 хиляди такива пациенти.
Детската прогерия е само косвено свързана с действителното остаряване. Това е заболяване от групата на ламинопатиите - заболявания, които се развиват на фона на проблем с производството на протеина ламин А. Ако той не е достатъчен или тялото произвежда „неправилния“ ламин А, тогава един от цял се развива списък от заболявания, който включва синдром на Hutchinson-Gilford.
Причината за детската прогерия е мутация в гена LMNA, който се намира на хромозома 1. Този ген кодира съединението преламин А, което произвежда протеина ламин А, който образува тънките ламини, покриващи вътрешната мембрана на ядрото. Необходим е за закрепване на всички видове молекули и вътрешни структури на ядрото. Ако липсва ламин А, вътрешната рамка на клетъчното ядро не може да се изгради, то не може да поддържа стабилност, което води до ускорено разрушаване на клетките и целия организъм. В допълнение, ламин А играе ключова роля в клетъчното делене. Регулира разграждането и възстановяването на клетъчното ядро. Не е трудно да си представим какво може да се случи, ако този протеин липсва или не е това, което трябва да бъде. Мутацията на гена LMNA води до образуването на "грешен" протеин - прогерин. Това е причината за ускореното „стареене” на децата.
По последни данни мутацията възниква при ранни стадииразвитието на ембриона и почти никога не се предава от родител на дете.
Преди няколко години учените откриха, че прогеринът се произвежда и от здрави клетки, но в значително по-малки количества, отколкото при синдрома на Hutchinson-Gilford. Освен това се оказа, че с възрастта прогеринът нормални клеткипораства. И това е единственото нещо, което наистина свързва детската прогерия и процеса на стареене.
Прогерията при възрастни е резултат от друга мутация в гена WRN. Този ген кодира протеин, необходим за поддържане на хромозомите в стабилно състояние и също така участва в процесите на клетъчно делене. Ако има мутация в гена WRN, структурата на хромозомите постоянно се променя. Честотата на спонтанните мутации се увеличава 10 пъти, докато способността на клетките да се делят намалява 3-5 пъти в сравнение със здравите клетки. Дължината на теломерите също намалява. И тези процеси вече са много близо до остаряването, което имаме предвид, когато гледаме възрастни хора на пейка.
Все още няма лек за прогерия. Лекарите се опитват да забавят бързото „стареене“ на такива пациенти. На деца със синдром на Hutchinson-Gilford се предписват лекарства с антиоксидантни свойства, витамин Е и др. Те се подлагат на коронарен артериален байпас в опит да се справят с усложненията в сърцето и кръвоносните съдове.
Лекарство, което може да удължи живота на пациент с детска прогерия с година и след най-добрият сценарийза 6-7 години, стана лонафарниб, лекарство, създадено за лечение на рак. За човек, чийто живот може да приключи на 13-17 години, това е значителен период. През 2012 г. беше установено, че приемът му води до подобряване на слуха, по-здрави кости, увеличаване на телесното тегло и повишена съдова гъвкавост. 6 години не са много, но сътворението на много ефективни лекарствазапочна с подобни постижения.
Окуражаващи резултати са получени и с рапамицин, правастатин и золедронат.
Необходима ли е профилактика на прогерията? Преди се смяташе, че не, тъй като хората с детска прогерия не живеят достатъчно дълго, за да могат да раждат деца, а възрастните пациенти развиват безплодие. Вярно, в последните годиниИма съобщения за жени с прогерия, раждащи здрави деца. Но това заболяване е толкова рядко, че вероятността от разпространение на мутации е изключително ниска.
Все още не е намерен лек за прогерия при възрастни. При такива пациенти се лекуват само тези тежки и многобройни заболявания, които ги атакуват.
„Гледайки това отпуснато лице, хлътнали очи и отпусната кожа, едва ли някой би си помислил, че това е дете. Това обаче е така.” Много хора знаят историята на 5-годишния Баязид Хосейн, който живее в Южен Бангладеш. Момчето страда от рядко генетично заболяване– прогерия, при която тялото и организмът остаряват осем пъти по-бързо от обикновено. Всичко започва с мускулна атрофия, дегенеративни процеси в зъбите, косата и ноктите, промени в костно-ставния апарат и този процес завършва с атеросклероза, инсулт и злокачествени тумори. Както виждаме, прогерията няма никак обнадеждаващи симптоми, които се превръщат в фатални опасни заболявания. Следователно такива пациенти винаги са изправени пред фатален изход. Но възможно ли е да се облекчат страданията им и дори да се удължи животът им? Или може би учените вече са на крачка от създаването на лек за това заболяване? Ще ви разкажем в днешната статия.
Синдром на Hutchinson при дете, Wikimedia
За първи път заболяване, при което тялото старее преждевременно, е идентифицирано и описано през 1889 г. от J. Hutchinson и независимо през 1897 г. от H. Guilford. Синдромът, който се проявява в детство.
Въпреки факта, че прогерията е доста рядко заболяване (само едно на 7 милиона новородени е диагностицирано с него), през цялата история на наблюдения на това заболяване в света вече са регистрирани повече от 150 случая. При раждането децата изглеждат абсолютно здрави, първите признаци на ускорено стареене започват да се появяват при бебета на възраст 10-24 месеца.
Причината за заболяването е мутация на гена LMNA, който произвежда протеина преламин А, който образува уникална протеинова мрежа - вътрешната рамка на ядрената обвивка. Резултатът е, че клетките губят способността си да се делят нормално.
Докато изследвали пациентите, генетиците открили и нарушения в възстановяването на ДНК (възстановителна функция), клониране на фибробласти (основните клетки на съединителната тъкан) и изчезването на подкожната тъкан.
По правило прогерията е ненаследствено заболяване и случаите на неговото развитие са редки, но има изключения. В няколко семейства такава мутация е регистрирана при деца братя и сестри - потомци на тясно свързани родители. И това показва възможността за автозомно рецесивен тип наследство, което се проявява при хора в зряла възраст. Между другото, това се случва на един на 200 000 души.
Още през 1904 г. немският лекар Ото Вернер забелязва драматични промени в външен види състояние при хора 14-18г. Той открива синдрома, който се свързва с внезапна загуба на тегло, забавен растеж, поява на сива коса и постепенно оплешивяване.
Всички тези трансформации на тийнейджър в старец са свързани с дефект в гена WRN (ATP-зависим хеликазен ген). Ролята на протеина WRN, който произвежда, е да поддържа геномната стабилност и да поддържа структурата и целостта на човешката ДНК. С течение на времето мутацията нарушава генната експресия, ДНК губи способността да се възстановява, което е причина за преждевременно стареене.
За разлика от малките пациенти, които не изостават, а в някои случаи дори надминават връстниците си умствено развитие, при възрастни се наблюдава обратен ефект, т.к прогерията започва да оказва пагубно влияние върху интелектуалните им способности.
Около 10% от пациентите до четиридесетгодишна възраст са изправени пред такива ужасни заболявания като саркома, рак на гърдата, астроцитом и меланом. Онкологията се развива на фона на захарен диабет и дисфункция на паращитовидните жлези. Ето защо средна продължителностПродължителността на живота на хората със синдром на Werner е 30-40 години.

В момента прогерията се счита за нелечимо заболяване. Животът на хората със синдром на Hutchinson (Hutchinson)-Gilford прекъсва на възраст 7-13 години, но има изолирани случаи, когато пациентите са живели до 20 или дори 27 години. И всичко това благодарение на някакъв вид лечение.
Специалистите от Progeria Research Foundation (PRF) и Бостънската детска болница обаче не бяха доволни от тази статистика. През 2012 г. те започнаха първите в света клинични изпитвания на лекарство, което може да помогне на бързо застаряващите деца. И както съобщава EurekAlert! , те успяха по този въпрос.
Проучването на пациенти с прогерия продължи 2,5 години. Учените поканиха за участие 28 деца от 16 различни страни, 75% от които бяха диагностицирани с болестта. Децата идваха в Бостън на всеки четири месеца и се подлагаха на пълно медицински преглед.
През целия период субектите се дават два пъти на ден специално лекарствофарнезилтрансферазен инхибитор (FTI), който първоначално е разработен за лечение на рак. Изследователският екип оцени динамиката на теглото, артериалната скованост (параметър за риска от инфаркт и инсулт) и костната скованост и плътност (параметър за риска от остеопороза).
В резултат на това всяко дете се почувства значително по-добре. Децата започнаха да наддават на тегло, имаше подобрения в структурата на костите и най-важното в сърдечно-съдовата система.
Според лекарите резултатите от това изследване са много обнадеждаващи. В бъдеще се планира да продължи изучаването на FTI лекарства и техния ефект, който ще даде Допълнителна информацияО сърдечно-съдови заболяванияи нормалния процес на стареене.
„Резултатите от това изпитание са обнадеждаващи за нашето семейство. Ние сме развълнувани и обнадеждени за бъдещето на Меган. Благодарни сме на Фондацията за изследване на прогерията и на всички лекари за техния ангажимент да помогнат на моята дъщеря и всички деца с прогерия“, каза Санди Найбър, майка на 12-годишната Меган, която участва в клиничното изпитване.

Повярвайте ми, никога не е твърде късно или в моя случай никога не е твърде рано да бъдете това, което искате да бъдете. Няма времева рамка - започнете, когато пожелаете. Можете да промените или да останете същите - за това няма правила. Можем да правим по-добри или по-лоши избори, надявам се вие да направите най-добрия. 
Този монолог е взет от филма на Дейвид Финчър „Странният случай с Бенджамин Бътън“, който е базиран на едноименния разказ на Франсис Скот Фицджералд.
От раждането си герой на това известна историябеше изгнаник, защото от ранна детска възраст имаше вид и здраве на 80-годишен старец: имаше бръчки по цялото тяло и атрофирали крака. въпреки това Времето тече, а Бенджамин, напротив, не остарява, а става по-млад. Много различни перипетии се случват на човека и, разбира се, любовта се случва в живота му.

В реалния живот такива чудеса не се случват и хората с прогерия никога не растат млади. Но въпреки болестта си, такива хора никога не престават да бъдат щастливи. По-специално, Леон Бота, южноафрикански художник, музикант и DJ, е известен на света не само със своите творческа дейност, както и това, че с ужасна болестуспя да живее до 26-годишна възраст.
Леон е диагностициран с прогерия на 4 години, но болестта не погубва живота му. Този човек обичаше да се наслаждава на всяка минута, въпреки че осъзнаваше, че предстоящата му смърт е неизбежна. Например през януари 2007 г. мъж организира първата си лична изложба в Дърбанвил, чиято тема е хип-хоп културата като начин на живот. Нека отбележим, че „младият“ мъж имаше няколко такива предавания.
Botha също се е занимавал с DJing и turntablism (вид DJing) и е участвал в известни клубове под псевдонима DJ Solarize. Освен това той си сътрудничи с южноафриканската група Die Antwoord и участва в техния видеоклип към песента Enter the Ninja.
Но, за съжаление, прогерията не щади никого. Следователно на 5 юни 2011 г. Бота почина от белодробна емболия - патологично състояниекогато част от кръвен съсирек (ембол), който се отделя от основното си място на образуване (често крак или ръка), се движи по кръвоносни съдовеи запушва лумена на белодробната артерия.
Днес учени от цял свят изучават това мистериозна болест. Те искат да го преместят от списъка на фаталните в списъка на неразрешимите. Заслужава да се отбележи, че науката вече е постигнала огромни резултати в тази посока. Остават обаче много въпроси, които трябва да бъдат разбрани, а именно: какви са приликите и разликите между специалните случаи на прогерия и нормалното стареене на тялото, как са свързани помежду си? генетични причиниСиндром на Werner и Hutchinson (Hutchinson)-Gilford и как да се противопоставим на ускореното стареене на тялото. Може би след известно време ще бъдат намерени отговори и специалистите ще могат да предотвратят развитието на болестта, като по този начин ще удължат живота на хората с прогерия.
Ако намерите грешка, моля, маркирайте част от текста и щракнете Ctrl+Enter.
Прогерията е синдром на преждевременно стареене, проявяващ се с характерни промени в кожата и вътрешните органи. Това е открита рядка генетична аномалия 1 човек на 4 милиона. В света има не повече от осемдесет наблюдавани случая на това заболяване. Етиопатогенетичните фактори на прогерията не са напълно проучени.
Има две морфологични форми на патология:
Терминът "прогерия" в превод от старогръцки означава "ранно стареене". Неестественото изчерпване на всички системи за поддържане на живота се дължи на генетична повреда. В същото време процесът на стареене се ускорява десетократно.
За синдрома на Hutchinson-GilfordДецата със забавено физическо развитие показват признаци на стареене: оплешивяване, бръчки, специфичен външен вид. Тялото им се променя значително: структурата на кожата е нарушена, вторичните полови белези липсват, вътрешните органи изостават в развитието си. След това бързо се развиват заболявания, свързани с възрастта: загуба на слуха, артроза-артрит, атеросклероза, инсулт или инфаркт, деминерализация на костите. Осемгодишно дете с това заболяване изглежда и се чувства на 80 години. В умственото развитие болните деца остават абсолютно адекватни. Техен интелектуално развитиене страда. Те рядко живеят след 13 години. Момчетата страдат от прогерия малко по-често от момичетата.

пример за развитие на дете с детска прогерия (синдром на Hutchinson-Gilford) от 1 година до 12 години
Синдром на Вернеробикновено започва да се проявява клинично при млади хора на възраст 16-20 години. Прогерия при възрастни – ускорено стареене с увреждане на всички системи и висок риск от рак различни локализации. Геномната нестабилност, която движи нормалния процес на стареене, води до различни патологични промени. Такива пациенти умират до 30-40-годишна възраст, като имат всички симптоми на дълбока старост.

пациент с прогерия при възрастни (синдром на Вернер) - преди началото на заболяването на 15 години и с развита форма на 48 години
Прогерията е нелечимо заболяване, което „отнема” детството на болните деца и ги „превръща” в истински старци. Редовно и адекватно здравеопазваневи позволява да забавите необратимия процес на стареене и да намалите тежестта клинични симптоми. За тази цел те използват лекарства, хранителни добавки, хирургични и физиотерапевтични техники.
Основната причина за прогерия е единична генетична мутация, чийто механизъм засега не е известен. Някои учени смятат, че истинската причинамутации се крият в наследствеността на родителите, други - в въздействието на радиацията върху ембриона по време на рентгенови лъчи на бременна жена.
 При синдрома на Werner процесът на възпроизвеждане на ДНК молекули е нарушен, а при синдрома на Hutchinson-Gilford се нарушава биосинтезата на протеина, който определя формата на клетъчните ядра.Генетичните нарушения правят клетките нестабилни, което води до стартиране на неочаквани механизми на стареене. Голям бройпротеинът се натрупва в клетките, които спират да се делят. В този случай черупката на ядрото става нестабилна и клетките на тялото стават неизползваеми и умират преждевременно. Мутацията води до производството на съкратен прогеринов протеин, който е нестабилен и бързо се разгражда в клетката. За разлика от целия протеин, той не се интегрира в ядрената ламина, която се намира под ядрената мембрана и участва в организацията на хроматина. Ядреният субстрат се разрушава, което води до сериозни проблеми. Прогеринът се натрупва в гладкомускулните клетки съдова стена. Дегенерацията на тези клетки е една от водещите прояви на заболяването.
При синдрома на Werner процесът на възпроизвеждане на ДНК молекули е нарушен, а при синдрома на Hutchinson-Gilford се нарушава биосинтезата на протеина, който определя формата на клетъчните ядра.Генетичните нарушения правят клетките нестабилни, което води до стартиране на неочаквани механизми на стареене. Голям бройпротеинът се натрупва в клетките, които спират да се делят. В този случай черупката на ядрото става нестабилна и клетките на тялото стават неизползваеми и умират преждевременно. Мутацията води до производството на съкратен прогеринов протеин, който е нестабилен и бързо се разгражда в клетката. За разлика от целия протеин, той не се интегрира в ядрената ламина, която се намира под ядрената мембрана и участва в организацията на хроматина. Ядреният субстрат се разрушава, което води до сериозни проблеми. Прогеринът се натрупва в гладкомускулните клетки съдова стена. Дегенерацията на тези клетки е една от водещите прояви на заболяването.
Прогерията при възрастни се унаследява по автозомно-рецесивен начин. При децата мутацията не се наследява, а се появява директно в тялото на пациента. Това не е изненадващо, тъй като носителите умират преди репродуктивна възраст.
Негенетични фактори, влияещи върху развитието на заболяването:
При раждането болното дете изглежда като нормално бебе. Клиничните признаци на прогерия се появяват още през първата година от живота. Някои деца се развиват правилно до 2-3 години, а след това започват да изостават от връстниците си по отношение на ръст и тегло. Децата с прогерия имат специфичен външен вид, тъй като признаците на заболяването са характерни и уникални. Всички пациенти са поразително подобни един на друг.

типични деца със синдром на Hutchinson-Gilford от различни семейства)

4-годишно момче с по-малко типична форма на синдром на Hutchinson-Gilford
Първо Клинични признациСиндромът на Вернер се появява на възраст 14-18 години. До пубертета пациентите се развиват нормално. Тогава те започват да изостават от връстниците си във физическо развитие, оплешивяват и посивяват. Кожата им изтънява, набръчква се и става нездравословно бледа. Ръцете и краката изглеждат много тънки поради атрофия на подкожната мастна тъкан и мускулите.

37-годишен мъж със синдром на Werner
След 30 години в тялото на пациентите се развиват следните патологични процеси:
Кожният епидермис е сплескан, влакната на съединителната тъкан са склерозирани, подкожна тъканатрофира и се замества частично съединителната тъкан. Ограничаването на пасивните движения в ставите на ръцете и краката се проявява чрез невъзможност за пълно огъване и разгъване на крайника. Това се дължи на цикатрициално стягане на сухожилията и болка.
До 40-годишна възраст пациентите развиват старчески заболявания: сърдечни проблеми, захарен диабет, чести фрактури на ръцете и краката, болки в ставите, доброкачествени и злокачествени кожни тумори, дисфункция на паращитовидните жлези. Рак, инфаркт и инсулт, вътрешни кръвоизливи са основните причини за смърт при прогерия.
Симптомите на патологията само приличат на процеса на нормално стареене. Признаци на стареене при прогерия са различни степениизрази или се появяват в различен ред. При естествено стареене растежът на ноктите се забавя, а при прогерия спира напълно. При по-възрастните хора веждите изтъняват след загуба на коса на главата, а при пациенти с прогерия е точно обратното.

Синдром на Hutchinson-Gilford

Синдром на Вернер
Диагностиката на прогерия не изисква специфични техники и изследвания. Външни знацизаболявания са толкова красноречиви, че диагнозата се поставя само по симптоми и данни визуална инспекция. Специалистите изучават лична и семейна история.
Показани са допълнителни изследвания за идентифициране съпътстващи заболявания. Пациентите са предписани общ анализкръв, нея биохимични изследвания, рентгенография на костно-ставния апарат, хистологично изследване на кожата, медико-генетично консултиране.
В момента няма панацея за прогерия. Всички лечения, които някога са били използвани, са се оказали неефективни. Лекари с помощ съвременни методиопитвайки се да спре болестта и да предотврати влошаването й. Пациентите се лекуват съвместно от специалисти в областта на ендокринологията, терапията и кардиологията.

За да облекчат състоянието на пациентите, лекарите предписват:
Провеждат се физиотерапевтични процедури за повлияване на схванати и схванати стави. На пациентите се предписва електрофореза, рефлексология, тренировъчна терапия, инфрачервени лъчи, водни процедури, калолечение, UHF терапия, магнитотерапия. Показан е при пациенти с прогерия правилното хранене, обогатен с витамини и микроелементи, умерен стрес от упражнения, дълги разходки свеж въздух, пълна почивка.
Бебетата се хранят през сонда със специални млечни формули, съдържащи добавки за наддаване на тегло. Млечните зъби се отстраняват, за да се освободи място постоянни зъби, които бързо избухват при болни деца. Специалистите наблюдават състоянието на сърдечно-съдовата система, което позволява ранно откриване на възникващи заболявания. хирургиясъщо е показан за пациенти със синдром ранно стареене. С ангиопластика или коронарен артериален байпасвъзстановяване на проходимостта на кръвоносните съдове.

Експерименталното лечение на възрастни със стволови клетки и инхибитори на фарнезилтрансферазата позволява възстановяване на подкожната мастна тъкан, общо тегло, намаляват чупливостта на костите. Прогнозата на заболяването винаги е неблагоприятна. Болните умират от остър коронарна недостатъчностили онкопатология. Предотвратяването на прогерия е невъзможно поради факта, че заболяването е генетичен характер. Терапията през целия живот може само да улесни и удължи живота на пациентите. Продължителните грижи, кардиологичните грижи и физиотерапията са основните направления в лечението на заболяването.Прогерия - нелечима патология, чието развитие не може да бъде спряно.

Двойствеността вълна-частица на светлината означава, че светлината едновременно има свойствата на непрекъснато електромагнитно...

Ролята на биологията е огромна в нашия свят. Въпреки че не е приоритетен предмет, повечето ученици и родители...
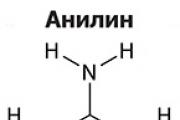
Амините са органични производни на амоняка, съдържащи NH2 аминогрупа и органичен радикал. Като цяло формулата...
Как да отговаряте на въпроси в част B. Втората част от работата по социални науки се състои от 7 задачи с кратък отговор. Част Б...
Формуляр TORG-15 се съставя в случай, когато по време на транспортиране, движение между и в склад, по време на съхранение...

Диетолозите казват, че за добро здраве и стройна фигура трябва да включите леки закуски в менюто си....

Вкусни мариновани моркови за зимата можете да приготвите в различни съдове, може да бъде дървена...

Когато ми останат няколко минути за приготвяне на закуска, се използват най-простите и бързи рецепти....

Изискана маса, украсена елха, дух от мандарини, разлят във всички стаи, скоро най-вълшебният празник -...

Всеки от нас многократно се е сблъсквал с финансови затруднения и трудни периоди в живота, когато Fortune...

Ако сте нови в магията, тогава ще ви бъде полезно да се запознаете със знаците, по които можете точно...

ТАЙНИТЕ НА СЪНИЩАТА Защо денят следва нощта? Какво е живот? Какво е смъртта и какво е сънят? Тези въпроси...

Основно значение: Която и версия на колодата на мадам Ленорман да вземем, определено можем да кажем, че това е една от...

Пример за коректна декларация за облагане на доходите през 2017 г., изтеглете безплатно в ексел новата актуална...

Ролята на биологията е огромна в нашия свят. Въпреки че не е сред приоритетните предмети, повечето ученици и...
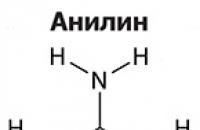
Амините са органични производни на амоняка, съдържащи NH2 аминогрупа и органичен радикал. Общо взето...