Испанска муха за двама – как влияят на либидото при жените и мъжете
Съдържание Биологично активна добавка на базата на екстракт, получен от бръмбар с муха (или муха...
Въпреки че синдромът на Rett е вродено заболяванеПрез първите шест месеца родителите може дори да не подозират, че детето им е зле. Поставянето на точна диагноза се усложнява допълнително от факта, че симптомите на това заболяване са подобни на тези на други заболявания. Междувременно навременното разпознаване на синдрома дава възможност за подобряване на качеството на живот на детето, въпреки че е невъзможно напълно да се излекува това генетично заболяване.
Синдромът на Rett или цереброатрофичната хиперамонемия е генетично дегенеративно заболяване, което засяга сивото вещество на мозъка. Проявява се като поредица от неврологични разстройства, които се изразяват в аутистично поведение, както и регресия на големи и фина моторика. Пациентите имат забавен растеж на ръцете, краката и главата, нарушения на стомашно-чревния тракт, сколиоза. Около 80% от децата страдат епилептични припадъци, приблизително 50% не могат да се движат самостоятелно.
Първите симптоми на заболяването се появяват, като правило, на възраст 4-6 месеца под формата на изоставане в развитието.Диагностицирането на заболяване на този етап е доста трудно, но с течение на времето заболяването прогресира по толкова характерен начин, че няма съмнение относно диагнозата. Тъй като синдромът на Рет е генетична патология, невъзможно е да се разболеете в зряла възраст: заболяването се диагностицира изключително при деца.
За първи път това заболяване е изследвано от австрийския педиатър Андреас Рет в средата на 20 век. Той описва повече от 30 свои пациенти с подобни симптоми. ДА СЕ характерни особеностиСиндромът на Rett класира регресията на умственото, умственото и физическо развитие, както и специални стереотипни движения на ръцете: стискане, изцеждане, преплитане на пръсти, имитация на миене на ръце и др. Интересно е, че идеята е да се отделят тези симптоми в индивидуално заболяванедойде в съзнанието на откривателя през 1954 г., докато диагнозата "синдром на Рет" се появи в медицината едва през 1983 г., след публикуването на трудовете на шведски учени по тази тема.
По принцип патологията се среща при момичета.Това се дължи на факта, че генът, в който причиняващи болестимутация, разположена на Х хромозомата. Жените имат 2 от тях, следователно, като имат 1 нормален ген в наличност, женските бебета са по-жизнеспособни. Мъжки плод, който има само една Х-хромозома с "счупен" ген, като правило умира в утробата. Редките случаи на раждане на момчета със синдром на Rett често са придружени от допълнителна аномалия - синдром на Klinefelter, тоест те са били носители на една допълнителна Х-хромозома.
Статистиката за разпространението на заболяването е доста висока: от 10-15 хиляди бебета 1 дете се ражда със синдром на Rett.
Синдромът на Rett започва да се изучава активно едва през последните 15-20 години. Причината за възникването му все още не е напълно изяснена, но се смята, че за всичко е виновна наследствеността. Това убедително доказва териториалният анализ на разпространението на патологията. По време на проучването в Италия, Норвегия, Унгария и Албания са открити няколко малки населени места, в които патологията е най-разпространена.
Въпреки доказаните наследствено предразположениекъм патологията, причините за синдрома на Rett все още не са проучени. Има няколко хипотези относно този механизъм. тази болест, но никой от тях не е единственият правилен.
Напълно възможно е всичките 3 фактора да повлияят на развитието на болестта или науката да открие нов в бъдеще, докато неизвестна причинапоява на синдром на Rett. Засега със сигурност се знае само едно: след раждането развитието на мозъка се забавя значително и до четиригодишна възраст при пациентите настъпват необратими морфологични промени в черепа, които водят до спиране на растежа, нарушаване на вътрешните органи, мускули атрофия и епилепсия.
Децата със синдром на Rett през първите месеци от живота си изглеждат напълно здрави. Тяхното развитие и телесни параметри са в рамките на нормалното. Единствените симптоми, които могат да се подозират за нещо нередно, са следните прояви:
До 4-5 месеца, когато децата започват активно да се преобръщат и да се опитват да пълзят, бебетата със синдром на Rett може да забележат изоставане в развитието. ярко тежки симптомипатологиите се появяват на шест месеца, въпреки че при някои деца заболяването дебютира на 1,5-2,5 години.
Интересното е, че при някои жени синдромът на Rett може да не се прояви клинично по никакъв начин и мутацията в гените се открива случайно: например, когато това заболяване се открие при техните дъщери.
Основните симптоми на синдрома на Rett са:
Характерен симптом на заболяването са стереотипните движения на ръцете, описани от А. Рет.Детето сякаш непрекъснато мие ръцете си, търка ги, стиска, пляска с пръсти, често дори хапе свитите си юмруци. Такива прояви абсолютно не са типични за изоставане в развитието или деменция, така че компетентен лекар веднага ще подозира, че детето има съответния синдром.
Заболяването е разделено на 4 последователни етапа.
| сцена | Възраст | Симптоми |
| Първо | 4 месеца - 2 години |
|
| Второ | 2–3 години |
|
| трето | 3–10 години |
|
| 4-ти | След 10г |
|
Периодите на относителна стабилност могат да бъдат заменени от фази на обостряне на заболяването. През такива периоди по-чести епилептични припадъци, поведенчески разстройства и влошаване общо благосъстояние. В четвъртия стадий на заболяването честотата на припадъците може значително да намалее, но децата често вече не могат да седят и ходят сами, освен това развиват много съпътстващи заболяванияпоради патологии на вътрешните органи.
Въпреки спирането на растежа и пълната неподвижност, пациентите със синдром на Rett преминават през всички етапи на пубертета навреме и могат да продължат да водят вегетативно съществуване в продължение на десетилетия.
Ако се появят симптоми, характерни за синдрома на Rett, трябва незабавно да покажете детето на психиатър.Няма нужда да се страхувате от този лекар: той няма да се регистрира без основателна причина и ако подозренията се потвърдят, навременната терапия може значително да подобри състоянието на бебето.
Психиатричната консултация започва с анамнеза. По време на разговор с родителите специалистът установява:
След разговора следва преглед на детето, по време на който специалистът оценява състоянието на мускулите, гръбначния стълб, както и растежа и пропорционалността на детето, по-специално наличието на хипотрофия на краката и ръцете. За точна диагноза лекарят често предписва допълнителни изследвания:
При липса на данни от тестове, синдромът на Rett може да бъде объркан с аутизъм.И двете състояния се характеризират с:
Възможно е да се разграничи едно заболяване от друго чрез диференциална диагноза, разработена през 1988 г. на Международната конференция за синдрома на Rett.
| знак | Синдром на Rett | RDA |
| Аутистични черти на възраст 6–18 месеца | Липсва | Често се появяват |
| стереотипи | Ритмични движения на двете ръце по средната линия на тялото | По-сложен и не в средната линия |
| Стереотипно манипулиране на обекти | Липсва | типичен |
| Моторика на багажника и походка | Прогресивна атаксия и апраксия (лоша координация) | Маниер, понякога грациозна стойка и походка |
| гърчове | Значително по-висока честота и полиморфизъм | Значително по-ниска честота и полиморфизъм |
| Респираторни нарушения, бруксизъм, забавяне на растежа на главата и крайниците | типичен | Липсва |
Използвайки диференциална диагноза, не само лекарите, но и самите родители могат да разграничат аутизма от синдрома на Rett.
На този етап от развитието на медицината синдромът на Rett е нелечимо заболяване.Въпреки това, с помощта на лекарства, рехабилитационни техники и специална диета е възможно да се подобри състоянието на детето, да се предотвратят сериозни деформации на тялото и да се подобри качеството на живот на пациента.
За подобряване общо състояниеЗа облекчаване на симптомите Вашият лекар може да предпише следните лекарства:
При тежка епилепсия антиконвулсантите може да не са ефективни. Често децата със синдром на Rett "надрастват" самите припадъци: до 10-годишна възраст пристъпите стават редки, а понякога изчезват напълно.
Желателно е приемът на лекарства да се комбинира с рехабилитационни мерки. Задължителни масажни сесии и ежедневно физиотерапиянасочени към коригиране на позата и предотвратяване на сколиоза. В допълнение, редовните упражнения под наблюдението на инструктор по тренировъчна терапия ви позволяват да научите детето си на двигателни умения. Масаж и лечебна гимнастикадобре се комбинира с лечебни сесии при хиропрактик, особено в по-напреднала възраст, когато се появяват признаци на сколиоза.
Музикалната терапия, по-специално методът на Томатис, е изключително ефективна при синдрома на Rett.Систематичното слушане на специално обработена музика насърчава нормализирането психо-емоционално състояниедеца и според родителите те са по-склонни да контактуват с други хора. В допълнение, такива класове могат да се провеждат в позната среда за детето - у дома.
Освен това, за да развиете умения за самообслужване и да се адаптирате към външния свят, можете да опитате следните техники:
Не забравяйте за редовните класове с дефектолози, психолози и логопеди, които често дават добър ефект в комбинация с приема на ноотропи. Също така, лечебните сесии с компетентни остеопати могат да помогнат на деца със синдром на Rett.
Малките деца с това заболяване трябва да участват в игри и развлекателни дейности възможно най-често: такова забавление допринася за общо развитиеи учи детето да взаимодейства ефективно с външния свят.
Тъй като децата със синдром на Rett често изпитват бърза загуба на тегло, те трябва специална диетас високо съдържаниемазнини. Менюто на такива пациенти трябва да съдържа витамини и фибри. Диетата трябва да бъде наситена висококалорични храни, включително специални смеси за наддаване на тегло.
Тези деца трябва да се хранят много по-често от здрави бебета. По правило лекарите препоръчват хранене на всеки 3 часа.Тази диета подобрява физическо състояниепациенти, предотвратяват изтощението и внезапната загуба на тегло.
В момента има активен научна работав зоната за търсене ефективно лечениетази болест. Има няколко хипотези по този въпрос и една от тях, включваща използването на инсулин като растежен фактор, вече се тества върху животни. По същия начин се тества и друга теория – за използването на стволови клетки при лечение. Като цяло учените са съгласни, че с течение на времето синдромът на Rett ще бъде окончателно победен, но времето за откриване на ново лекарство все още не е обявено.
Въпреки тежестта на заболяването, прогнозата за продължителността на живота е доста оптимистична. Възрастните жени със синдром на Rett често живеят до 40-те години и след това. Най-честите причини за смърт са:
Прогнозата за децата от мъжки пол е разочароваща: момчетата със синдром на Rett често не живеят повече от 2 години, умирайки от тежка енцефалопатия.
Синдромът на Рет е сериозно и все пак нелечимо заболяване. Въпреки това, родителите могат да облекчат състоянието на децата и да увеличат продължителността на живота им, като открият болестта навреме и спазват всички предписания на лекарите. В същото време всяка година ще ги доближава до времето, когато учените открият ефективно лекарствоот това заболяване и ще даде надежда за пълно възстановяване.
Какво представлява синдромът на Турет, защо може да се появи, какви симптоми се появяват по време на атаки? Диагностика и лечение на генерализиран тик различни методи. Възможно ли е да се предотврати наследствено заболяване?
Съдържанието на статията:
Синдромът на Gilles de la Tourette (генерализиран тик) е наследствено заболяване, чиито първи симптоми започват да се появяват в детството. Генетичното заболяване се причинява от дисфункционално функциониране на нервната система, поради което пациентът развива множество двигателни тикове, придружени от гласова проява.

Генерализираният тик е доста рядък - в момента се диагностицира при 0,05% от населението. Етиология на заболяването съвременна медицинане е напълно проучен. Специфичният ген, който причинява нервно-психични разстройства, все още не е идентифициран, но е установено, че вероятността за предаване на синдрома на Турет по наследство е 50%, но това не означава, че болестта непременно ще се прояви. Момчетата боледуват по-често от момичетата, с около 70-80%.
Външните фактори не могат да провокират развитието на болестта, но те утежняват нейния ход. ДА СЕ външни факторисе отнасят инфекциозни заболявания, по-специално причинени от въвеждането на стрептококова инфекция. Това се дължи на увреждане на мозъка от патогенна микрофлора от този тип.
При хора със синдром на Турет най-голямото числонегативните прояви са фиксирани в юношеството, до 20-годишна възраст при 90% от пациентите заболяването регресира. При 10% от пациентите състоянието се влошава и може да причини увреждане и да бъде причина за увреждане.
Синдромът на Турет не винаги се диагностицира с множество тикове. Болестта изисква много диагностични мерки. Подобни прояви се наричат туретизми и лечението се провежда по други терапевтични схеми.

Увеличете вероятността от синдром на Турет:

Тиковете могат да бъдат класифицирани като двигателни - прости и сложни, вокални. В простите двигателни мускули участват мускули от една и съща група. Те могат да се характеризират като свиване на устните, щракане и скърцане със зъби, мигане или свиване на веждите, неволно махане с ръце или крака.
Сложните тикове са движения, при които се появяват изразени гримаси, неволно подскачане, докосване на някои части на тялото, удряне на главата в предмети, конвулсии, поради които пациентът може да се нарани. Характеристики на тиковете: монотонност, липса на ритъм, осъзнаване на случващото се, възможност за краткотрайно волево потискане. В бъдеще пациентите могат да обяснят движенията си като усещане за пясък в очите, тежест и напрежение в крайниците, външен вид чуждо тялоили силен звук под свода на черепа и др.
Вокалните тикове могат да се изразят като звукови звуци - сумтене, кашляне, използване на цели фрази или изречения. Има такива видове вокални тикове:


Необходимо е да се наблюдават в динамика промените в изследванията на урината - според Zemnitsky и общи, кръвни - подробни, биохимични. Преминете специфични тестове на урината за метаболити. Не забравяйте да предпишете хардуерни изследвания: кардиограма, невросонография, CT или MRI, електроневрография - оценка на скоростта на нервните импулси.
Как да се лекува синдром на Турет се определя след сравняване на резултатите изчерпателни проучвания. Показания за тях са проявите на симптомокомплекса през годината.
Синдромът на Турет 1-2 степен не изисква употребата на лекарства, но това не означава, че пациентът ще може сам да се справи с болестта. В този случай е необходимо да се консултирате с психолог или психотерапевт, за да обясните на родителите особеностите на поведението при отглеждане на болно дете. 3-4 степен на проява на генерализиран тик изисква продължителна и постоянна употреба медицински препарати.

Необходимо:
Много е важно да се разбере, че вокалните тикове не трябва да бъдат порицавани, тъй като това може да влоши тежестта на симптомите.
Деца със синдром на Турет изискват повишено вниманиеи специални грижи.

Терапевтичните ефекти се определят индивидуално за всеки пациент. Могат да бъдат включени следните техники:

IN стационарни условияЗа лечение на пациенти могат да се използват следните лекарства:
Лечението се допълва от назначаването на витаминно-минерален комплекс с преобладаване на витамини от група В и високо съдържаниемагнезий.

Въпреки това, след 4-8 години, признаците на заболяването се появяват отново и все още не е постигната стабилна ремисия. Освен това операциите са много сериозни, рискът от развитие странични ефектимного висок. Операцията на мозъка може да се извършва само от висококвалифицирани хирурзи.
Всички лекарства, използвани за лечение на генерализирани тикове, имат изразени странични ефекти. Тези лекарства се предлагат само по лекарско предписание. В никакъв случай не трябва да се самолекувате - това може да доведе до значително влошаване на състоянието.

Има обаче мерки, които могат да помогнат за намаляване на риска от симптоми:
Синдромът на Турет не влияе върху продължителността на живота.
Как да лекувате синдрома на Турет - вижте видеото:
- генетично хетерогенно наследствено заболяване, характеризиращо се с различни метаболитни нарушения и образуването на компоненти на централната нервна система. Симптомите на тази патология, като правило, се появяват дори в ранна детска възраст, те включват мускулна хипотония, проблеми с храненето и психомоторна изостаналост. При по-нататъшно прогресиранезаболявания възникват епилептични припадъци, хиперкинеза, респираторни нарушения. Диагнозата на синдрома на Лий се извършва въз основа на данни за текущото състояние на пациента, ядрено-магнитен резонанс и молекулярно-генетични анализи. специфично лечениетази патология не съществува, симптоматичната терапия само леко забавя прогресията на заболяването.

Синдромът на Leigh (подостра некротизираща енцефаломиелопатия) е наследствено невродегенеративно заболяване на централната нервна система, характеризиращо се с ранен старти стабилното прогресиране на неврологичните разстройства. Първо дадено състояниее описано през 1951 г. от английския психиатър Денис Лий, който го определя като наследствен вариантенцефаломиелопатия. По-нататъшни проучвания показват, че синдромът на Leigh е изключително хетерогенно състояние по отношение на етиологията - причинява се от дефекти в много гени, разположени върху автозоми, X хромозома и митохондриална ДНК. Поради тази причина механизмът на унаследяване на заболяването може да бъде (в зависимост от естеството на мутацията) автозомно-рецесивен, свързан с пола или митохондриален. Поради разнообразието от генетични дефекти, които причиняват синдрома на Leigh, разпределението на пола на това състояние също се различава, но според много генетици като цяло може да се счита, че то засяга еднакво както момчетата, така и момичетата. Честотата е приблизително 1 случай на 34-36 хиляди новородени.
Причините за развитието на синдрома на Leigh могат да бъдат мутации на широк спектър от гени, разположени на различни хромозоми. Въпреки това, патогенезата на това състояние е приблизително сходна при различни форми на патология и най-често се свързва с нарушение на процесите на клетъчно дишане и функционирането на дихателната верига на митохондриите. По отношение на някои форми на синдрома на Leigh също се забелязва нарушение на функционирането на комплекса пируват дехидрогеназа. Нарушаването на протеиновата структура на дихателната верига на митохондриите води до недостатъчен синтез на АТФ, който е основният източник на енергия във всички клетки на тялото. Невроните и невроглиалните клетки са особено чувствителни към липса на енергия, което причинява развитието различни нарушенияоще от детството. Класификацията на всички генетични дефекти при синдрома на Leigh се основава на това кой компонент от дихателната верига (която е протеинов комплекс) на митохондриите е увреден в резултат на мутацията.
Като отделен вариант на синдрома на Leigh често се посочва форма на заболяването поради мутации в гена PDHA1, който се намира на X хромозомата. В резултат на това унаследяването на този вид патология е свързано с пола - почти изключително момчетата са болни, докато жените са носители на патологични гени. Митохондриалният начин на наследяване на синдрома на Leigh също има много характеристики. Предаването на патологични гени става от майката на потомството и продължава само през женска линия. Тъй като всяка митохондрия има своя собствена ДНК молекула, както „здрави“, така и „болни“ органели присъстват едновременно в клетката и по време на клетъчното делене (включително мейозата в процеса на образуване на яйцеклетката) разпределението на болните гени не е същото. Жените с относително малък процент "болни" митохондрии в клетките си могат да бъдат фенотипно здрави, но ги предават на потомството си. Невъзможно е точно да се предскаже колко анормални митохондрии ще получи дете от такива носители, така че вероятността от развитие на синдром на Leigh при децата на тези жени е несигурна.
Проявите на синдрома на Leigh обикновено се появяват през първата година от живота на детето, понякога те могат да бъдат записани на възраст 2-5 години, в редки случаиразвитието на болестта започва в юношеска възраст. Обикновено първата проява на патологията е сънливост или, обратно, свръхвъзбудимостдете, при кърмачетаима нарушение на храненето, липса на телесно тегло. В бъдеще синдромът на Лий води до забавяне на психофизическото развитие, а при по-големи деца - до постепенна загуба на вече придобити умения. Сред другите неврологични симптоми на заболяването, пареза, тремор на крайниците, нарушена координация на движението, увреждане на периферни нерви, намалени сухожилни рефлекси. В бъдеще могат да се регистрират клонични конвулсии и епилептични припадъци.
Поради липсата на енергия поради синдрома на Leigh страда не само нервната система, но и други органи с висока консумация на АТФ. В повечето случаи болните деца имат мускулна хипотония и слабост. Болестта засяга и черния дроб, орган с много висока консумация на енергия. При пациенти със синдром на Leigh често се открива уголемяване на черния дроб, жълтеница и понякога хепатолиенален синдром. С напредването на патологията се появяват нарушения на дишането - затруднява се, понякога придобива характера на дишането на Чейн-Стокс. Редица пациенти в крайна сметка развиват миокардна дистрофия.
Синдромът на Лий има прогресивен курс. В крайните етапи се наблюдава увреждане на органите на зрението, което се проявява чрез нистагъм, нарушено цветоусещане и страбизъм. В крайна сметка може да настъпи атрофия на зрителния нерв и пълна слепота. Мускулната хипотония и хипорефлексия се заменят със спастично мускулно напрежение и повишени сухожилни рефлекси. 2-7 години след появата на първите симптоми на синдрома на Leigh, рязък спадтелесно тегло, горните прояви се увеличават рязко, смъртпоради дихателна или сърдечно-съдова недостатъчност.
За да се определи наличието на синдром на Leigh, ядрено-магнитен резонанс на мозъка, електроневромиография, изследване наследствена история, молекулярно-генетични анализи. При преглед характерни неврологични симптоми, тремор на крайниците, изоставане в психофизическото развитие, при кърмачета - липса на телесно тегло. Магнитно-резонансното изображение на мозъка разкрива симетрични промени в продълговатия мозък, таламуса и моста, понякога подобни промени могат да бъдат регистрирани в гръбначен мозък. Най-добрите резултати при диагностицирането на синдрома на Leigh с помощта на MRI се получават при използване на режими T2W и FLAIR.
В случаите, когато има признаци на увреждане на периферните нерви и мускули, се извършва електроневромиография за диагностициране на синдрома на Leigh. При това заболяване основният и най общ резултат ENMR става забавяне на скоростта на преминаване нервен импулскоето показва демиелинизация на нервите. Проучването на наследствената история е информативно в случай на автозомно-рецесивни форми на заболяването; семеен характерпатологията е трудна. Молекулярно-генетичната диагностика се използва широко само по отношение на някои форми на синдром на Leigh (поради мутации в BCS1L, SURF1 и някои други гени).
Няма специфично лечение за тази патология, използва се симптоматична терапия: антиконвулсанти и ноотропи, лекарства за подобряване мозъчно кръвообращение. Важна роляпри лечението на синдрома на Leigh играе назначаването на витамини, които служат като кофактори на ензимите на дихателната верига на митохондриите - B1, B6, Q10. Редовният им прием може до известна степен да забави прогресията на заболяването и да намали тежестта на симптомите. Въпреки това, въпреки всички предприети терапевтични мерки, 80% от пациентите умират 2-7 години след регистриране на първите прояви на патологията.
Прогнозата на синдрома на Leigh е изключително неблагоприятна, тъй като повечето пациенти умират в рамките на няколко години след началото на заболяването. Симптоматично лечениеможе донякъде да забави прогресията на патологията и да намали тежестта на проявите, но не осигурява пълно подобрение. В повечето случаи, дори година или две преди смъртта, пациентът става напълно инвалидизиран поради неврологични, респираторни и метаболитни нарушения. Причината за смъртта при синдром на Leigh най-често е сърдечно-съдова или дихателна недостатъчност. Профилактиката на това заболяване се извършва в рамките на медицинско генетично консултиране на родителите преди зачеването на дете.
Синдромът на Leigh (подостра некротизираща енцефалопатия) е наследствено заболяване, което се проявява с неврометаболитен синдром, засягащ централната нервна система. Най-често това заболяване засяга деца под тригодишна възраст, много по-рядко се среща при юноши и възрастни.
Болестта на Лий може да бъде остра, подостра или хроничен ход. Показаните симптоми са достатъчно изразителни, за да се диагностицира синдромът ранни стадии. Разпространението на това заболяване зависи от наследствения фактор, ако в семейството има болен човек, тогава вероятността от предаване на болестта на деца е доста висока.
Подостра некротизираща енцефаломиопатия се развива поради липса на ензими, отговорни за образуването енергиен метаболизъмв организма.
Липсата на ензими възниква на фона на анормален метаболизъм на пирогроздена киселина и дефекти в миграцията на електрони в дихателните вериги. Синдромът на Leigh възниква и поради следните причини:
Синдромът на Leigh най-често се характеризира с подостро или хронично протичане, което е фатално. Много по-рядко  заболяването протича под формата на остра енцефалопатия.
заболяването протича под формата на остра енцефалопатия.
При бързата форма на развитие на заболяването възниква апоплексия на дихателния център, което води до смърт.
Съществуват меки формипротичане на заболяването, придружено от забавяне на психомоторната онтогенеза.
При всяка форма на заболяването курсът му е променлив. След бързо клинично влошаване настъпват краткотрайни подобрения.
Всяка форма на болестта на Лия се характеризира със собствена клинична картина. Подострата форма на енцефаломиопатия се проявява със следните симптоми:
При бърза и прогресивна форма на хода на синдрома се появяват следните симптоми:
Общ клинична характеристиказа всички форми на синдром на Leigh:

За да установите диагнозата на Лея, приложете следните методиизследвания и се правят следните анализи:

В допълнение към горните диагностични методи се провеждат и спомагателни изследвания:
 Лечението на некротизираща енцефалопатия не винаги е възможно. Ако заболяването прогресира до хронична форма, тогава смъртта е неизбежна.
Лечението на некротизираща енцефалопатия не винаги е възможно. Ако заболяването прогресира до хронична форма, тогава смъртта е неизбежна.
Целта на лечението е да се подобри състоянието на централната нервна система. За това се предписват лекарства, които включват витамин В1. Предписват се и антибиотици, най-добрите от които са ампицилин и биотин. При лекарствена терапиянеобходимо е да спазвате диета, при която да приемате не повече от 1 грам протеин на ден.
В риск са хората, които имат наследствена предразположеност към това заболяване и деца под тригодишна възраст. Ако не е предоставена медицински грижи, болестта свършва фатален. Средна продължителностживотът от момента на заболяването е пет години.
Профилактиката на заболяването се състои в навременното лечение на заболявания на централната нервна система и заболявания, които засягат централната нервна система. Също така е необходимо да имате балансирана диета и здравословен начин на животживот.
Към днешна дата, първият известен ефективен методпредотвратяване на синдрома на Leigh, който се състои в раждането на дете "от трима родители", използвайки митохондрии от донорни клетки.
Ефикасност този методпревенцията е доказана в края на 2016 г. и досега почти не се използва в практиката.

Съдържание Биологично активна добавка на базата на екстракт, получен от бръмбар с муха (или муха...

Екатерина МиримановаСистема минус 60. Революция Система минус 60 с Екатерина Мириманова"Система минус 60. Революция"...

Тежестта и подуването са причини както за обикновено преяждане, така и за по-сериозни проблеми с храносмилателния тракт....

Втората кръвна група, Rh-отрицателна, се появи преди много години, когато човек престана да бъде ловец и ...

ПСИХОЛОГИЧЕСКИ АСПЕКТИ НА ФЕНОМЕНА АНОРЕКСИЯ (ЕКСПЕРИМЕНТАЛНО ИЗСЛЕДВАНЕ) Т. В. Тарасова, Е. В. Арсентьева В...

Съдържание Поради факта, че кожата в тази област е тънка, тя е по-склонна към появата на различни видове петна. Зачервяване под...

vseslav сб, 17.10.2015 - 20:50 Станция Василеостровская е една от най-старите станции...

Най-често козата гъба (Suillus bovinus) в ежедневната реч на гъбарите се нарича коза. Тази тръбна...

Всеки човек поне веднъж в живота си е имал мечти. Мнозина не им придават никакво значение, но някои ...

Дедал (кратер). Диаметър: 93 км Дълбочина: 3 км (снимка на НАСА) Луната привлича вниманието на хората от древни времена. Във II...

Съединените щати празнуват Деня на президента на третия понеделник на февруари. До 70-те години празненствата са били насрочени за...

В деня на последното обаждане искам да кажа само една дума на моя любим главен учител: „Благодаря ви“. Но колко е...

Динята се счита за вкусно и здравословно зрънце, семената й са особено полезни. Малък, тъмнокафяв или...

„Развитие на речта при деца на 6 години“ Дете не се ражда с установена реч. Не е възможно да се даде категоричен отговор на...

Екатерина Мириманова Система минус 60. Революция Система минус 60 с Екатерина Мириманова "Система минус 60 ....
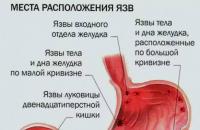
Тежестта и подуването са причини както за обикновеното преяждане, така и за по-сериозни храносмилателни проблеми.