What is possible and not possible during the Nativity Fast?
In 2018, the Nativity Fast will begin on November 28. During this period, Orthodox believers prepare to celebrate Christmas...
True or primary polycythemia (polycythemia vera) is a chronic tumor disease circulatory system person. Expressed in sharp jump level of red blood cells in human blood without damage internal organs. The blood becomes more viscous and thick, which leads to a slowdown in blood flow and, as a result, to the formation of blood clots and hypoxia, which affects the erythroblastic germ to a greater extent. This disease was first described by Vaquez in 1892, which is why it is also called Vaquez's disease.
This disease usually affects older and older people, and men are more often affected. Children and young people get sick much less frequently, but their illness is more severe. The real reasons diseases have not been established, but it has been proven genetic predisposition to this illness.
The disease develops slowly; for some time the patient may not even suspect that he has the disease, since early stages there are no signs. Then, as the disease progresses, patients begin to experience dizziness, an incomprehensible feeling of the skull being full, and constant headaches.
In the advanced stage of the disease, the following clinical symptoms are observed:

Patients also complain about fatigue, visual impairment, hearing impairment, mood instability and general lethargy. In addition, the disease is associated with high blood pressure, severe weight loss, frequent colds and bronchitis.
The diagnosis is made after a full assessment of the clinical picture, analysis of changes in the blood and examination bone marrow. The following indicators are taken into account to confirm the presence of Vaquez disease.
The level of red blood cells and hemoglobin is significantly increased; on the contrary, it is decreased and is less than one. A high red blood cell count leads to an increase in the total blood volume in the body (up to 2 times), and the hematocrit also changes (up to 65%).
Leukocytes are increased 1.5–2 times, neutrophils are growing (up to 80%). The number of platelets increases significantly (more than 400*109 per liter). Blood viscosity is significantly increased, red blood cells settle no more than 2 millimeters per hour. The rate is also rapidly increasing uric acid.
Using trepanobiopsy, a bone marrow puncture is taken. Histological examination of the punctate reveals an increase in megakaryocytes. At the same time, bone marrow cells retain the ability to mature at the same level.
There is also a need to produce differential diagnosis with secondary erythrocytosis.
Vaquez's disease tends to occur in the form of a protracted chronic process. The patient's life is at risk increased risk bleeding and blood clots.
The main direction of treatment is to combat thrombosis and reduce blood viscosity, which depends on the level of red blood cells. For this purpose, bloodletting and chemotherapy are used.
Normalizes the level of hemoglobin and red blood cells by reducing the patient's blood volume. Volume – up to 500 milliliters per procedure. Such procedures are carried out under the supervision of a hematologist once every two days until hemoglobin (140–150) and hematocrit (45%) decrease within normal limits. This method It stimulates the bone marrow well and relieves skin itching. In most cases, bloodletting is used in combination with chemotherapy.
Used to inhibit the recovery of red blood cells and platelets. For this purpose they are used pharmacological preparations different groups: imiphos, myelobromol, myelosan, chlorambucil, and also use hydroxyurea and pipobroman.
Polycythemia vera(erythremia, Vaquez's disease, polycythemia rubra) - IP is a chronic neoplastic myeloproliferative disease with stem cell damage, proliferation of three hematopoietic lineages, increased formation of red blood cells and, to a lesser extent, leukocytes and platelets. At a certain stage of the disease, myeloid metaplasia of the spleen occurs.
The incidence of polycythemia vera is approximately 1 random per 100 thousand population per year and in last years has an undoubted tendency to increase. Men get sick slightly more often than women (1.2:1). Average age sick people are 60 years old, patients under 40 years old make up only 5%.
Etiopathogenesis. Polycythemia vera is a clonal neoplastic disease, which is based on the transformation of a hematopoietic stem cell. Since malignant transformation occurs at the level of a pluripotent stem cell, all three lineages of hematopoiesis are involved in the process. In patients suffering from PV, there is an increased content of CFU-GEMM (colony-forming units - granulocytic, erythroid, macrophage and megakaryocyte) - progenitor cells close to a pluripotent stem cell. In cell culture, active proliferation of these cells occurs in the absence of erythropoietin. Low serum erythropoietin levels are a specific sign of PV. In the bone marrow, hyperplasia of predominantly erythroid cells, as well as granulocytic and megakaryocytic lineages, is observed. A characteristic feature is the presence of clusters of polymorphic megakaryocytes (from small to giant). Myelofibrosis is rarely observed at the time of diagnosis, but manifests itself clearly with a long course of the disease. Gradually there is an increase in the number of reticulin and collagen fibers, myelofibrosis develops and myelopoiesis is reduced. The mass of circulating erythrocytes (MCE) increases, hematocrit increases, blood viscosity increases (there is a significant increase in hemoglobin content in the blood (from 180 g/l and above), red blood cells (from 6.6 x 10 12 /l) and hematocrit (from 55 % and higher). These factors, along with thrombocytosis, lead to impaired microcirculation and thromboembolic complications. In parallel, myeloid metaplasia of the spleen is associated. In IP there is no specific cytogenetic marker, however, in a significant number of patients with IP in the stage of development of myelofibrosis, chromosomal anomalies.
Clinical picture changes with the course of the disease and is determined mainly by the stage of the disease. In the domestic literature, it is customary to distinguish four stages of IP, which reflect the pathological processes occurring in the bone marrow and spleen of patients
Stages:
I - initial, low-symptomatic (5 years or more):
the spleen is not palpable
moderate erythrocytosis
moderate plethora
in the bone marrow panmyelosis
Vascular and thrombotic complications are possible, but not common
External manifestations of the disease are plethora, acrocyanosis, erythromelalgia (burning pain, paresthesia in the fingertips) and itchy skin after washing. An increase in MCE and, consequently, in circulating blood volume leads to arterial hypertension. If the patient has previously suffered from hypertension, then the blood pressure level increases, and antihypertensive therapy becomes ineffective. Manifestations are getting worse coronary disease heart, cerebral atherosclerosis. Since MCE increases gradually, plethora, an increase in the number of red blood cells and hemoglobin, signs of microcirculation disorder in a number of patients appear 2-4 years before the diagnosis is made.
II – erythremic, expanded (10-15 years):
A. Without myeloid metaplasia of the spleen
general condition is disturbed
pronounced plethora (Hb 200 g/l or more)
thrombotic complications (stroke, myocardial infarction, necrosis of the fingertips)
panmyelosis
erythromelalgia (pain in the limbs and bones)
In the picture of peripheral blood, in addition to erythrocytosis, neutrophilia is often present with a shift of the leukocyte formula to the left to single myelocytes, as well as basophilia and thrombocytosis. In the bone marrow, total three-line hyperplasia with pronounced megakaryocytosis is detected, and reticulin myelofibrosis is possible. But at this stage of the disease there is still no myeloid metaplasia of the spleen (MMS), and the observed splenomegaly is due to increased sequestration of erythrocytes and platelets. Vascular complications are more frequent and severe than in the first stage of the disease. In the pathogenesis of thrombosis, an important role is played by an increase in MCE, leading to an increase in blood viscosity and a slowdown in blood flow, thrombocytosis, as well as dysfunction of the endothelium. Ischemia associated with impaired arterial blood flow is observed in 24-43% of patients. Thrombosis of cerebral vessels, coronary and arteries supplying blood to the abdominal organs predominates. Venous thrombosis is detected in 25-30% of patients and is the cause of death in approximately a third of patients suffering from PV. Thrombosis of the veins of the portal system and mesenteric veins is not uncommon. In a number of patients, it is thrombotic complications that become a manifestation of IP. Polycythemia vera may be accompanied by hemorrhagic syndrome: frequent nosebleeds and bleeding after tooth extraction. Hypocoagulation is based on a slowdown in the conversion of fibrinogen to fibrin, which occurs in proportion to the increase in hematocrit, and a violation of blood clot retraction. Erosion and ulcers of the stomach and duodenum considered as visceral complications of IP.
B. With myeloid metaplasia of the spleen (MMS).
hepatosplenomegaly
plethora is moderately expressed
panmyelosis
increased bleeding
thrombotic complications
Splenomegaly increases, the number of leukocytes increases, the shift of the leukocyte formula to the left becomes more pronounced. In the bone marrow - panmyelosis; Reticulin and focal collagen myelofibrosis gradually develops. The number of red blood cells and platelets decreases somewhat due to their increased destruction in the spleen, as well as the gradual replacement of hematopoietic tissue with fibrous tissue. At this stage, stabilization of the patient’s condition may be observed, the level of hemoglobin, red blood cells and platelets approaches normal without therapeutic measures.
III - anemic:
anemic sm (even pancytopenia)
severe myelofibrosis
liver, spleen enlarged
In the bone marrow, collagen myelofibrosis increases and myelopoiesis decreases. The hemogram shows anemia, thrombocytopenia, and pancytopenia. The clinical picture of the disease may include anemic and hemorrhagic syndrome s, splenomegaly and cachexia increase. The outcome of the disease may be transformation into acute leukemia and myelodysplastic syndrome (MDS).
Diagnostics. Currently, the criteria developed by the American Polycythemia Vera Study Group (PVSG) are used to establish the diagnosis of polycythemia vera. You-
1) an increase in the mass of circulating red blood cells (more than 36 ml/kg for men and more than 32 ml/kg for women);
2) normal saturation of arterial blood with oxygen (pO2 more than 92%);
3) splenomegaly.
1) thrombocytosis (platelet count more than 400 x 10 9 /l);
2) leukocytosis (the number of leukocytes is more than 12 x 10 9 /lb without signs of infection);
3) activity alkaline phosphatase(neutrophils above 100 units in the absence of fever or infection);
4) high content vitamin B12 (more than 900 pg/ml).
The diagnosis of IP is considered reliable if the patient has all three signs of category A, or if the first and second signs of category A and any two signs of category B are present.
Currently, the most important diagnostic sign is the characteristic histological picture of the bone marrow; hyperplasia of cells of erythroid, granulocytic and megakaryocyte lineages with a predominance of erythroid, accumulations of polymorphic megakaryocytes (from small to giant). Myelofibrosis is rarely observed at the time of diagnosis, but becomes distinct with a long course of the disease.
At stage I, polycythemia vera, characterized by isolated erythrocytosis, must be differentiated from secondary erythrocytosis, which is a response to any pathological process in the body and can be either true or relative.
Relative erythrocytosis is a consequence of hemoconcentration, that is, MCE is normal, but the plasma volume is reduced, which is observed with dehydration of the body (for example, taking diuretics, polyuria in patients with diabetes, vomiting and diarrhea), loss of a large amount of plasma due to burns.
True secondary erythrocytosis (MCE is increased, hematocrit is increased) is caused by increased production of erythropoietin. The latter is compensatory in nature and is caused by tissue hypoxia in people living at significant altitudes above sea level, in patients with pathologies of the cardiovascular and respiratory systems, and in smokers. This category also includes patients with hereditary hemoglobinopathies, characterized by an increased affinity of hemoglobin for oxygen, of which a smaller amount is released in the body tissues. Inadequate production of erythropoietin is observed in kidney diseases (hydronephrosis, vascular pathology, cysts, tumors, congenital anomalies), hepatocellular cancer, large uterine fibroids. An essential differential diagnostic feature is the level of serum erythropoietin.
Treatment. In the initial stages of the disease, it is recommended to use bloodletting, which significantly alleviates the manifestations of plethoric syndrome. The method of choice for reducing hematocrit (and hemoglobin to normal values) is bloodletting (exfusion), which is recommended if the hematocrit exceeds 0.54. The goal of treatment is a hematocrit of less than 0.42 for women and 0.45 for men. In modern conditions, bloodletting can be replaced by erythrocytepheresis. In addition, to facilitate bloodletting and prevent thrombotic complications, patients are given courses of disaggregant therapy (aspirin, rheopolyglucin, etc.). Choosing a treatment method at advanced stage II of IP is perhaps the most difficult task. In addition to erythrocytosis, patients have leukocytosis and thrombocytosis, and the latter can reach very high numbers. Some patients have already suffered some kind of thrombotic complications, and exfusions increase the risk of thrombosis.
When individualizing therapy, the age of the patients should be taken into account. This is the treatment of patients under 50 years of age, without a history of thrombotic complications and severe hyperthrombocytosis (< 1000,0 х 10 9 /л) может быть ограничено только кровопусканиями в сочетании с терапией аспирином (или без него) в дозе 100-375 мг в день.
For patients over 70 years of age, with a history of thrombotic complications and severe hyperthrombocytosis, therapy with myelosuppressive drugs is indicated. Patients 50 to 70 years of age without thrombotic complications or severe hyperthrombocytosis can be treated with myelosuppressive agents or phlebotomy, although the latter treatment may increase the risk of thrombotic complications.
Currently, in addition to bloodletting and antiplatelet agents, hydroxyurea and alpha-interferon are mainly used for the treatment of PV, less often busulfan, and anagrelide is used abroad. Hydroxyurea may be the drug of choice in patients with PV with severe leukocytosis and thrombocytosis. But for the sick young the use of hydroxyurea is limited by its mutagenic and leukemic effects. In addition to hydroxyurea, interferon-alpha is widely used in the treatment of PV. Firstly, IF-a suppresses pathological proliferation quite well and does not have a leukemic effect. Secondly, like hydroxyurea, it significantly reduces the production of platelets and white blood cells. Special attention deserves the ability of IF-a to eliminate skin itching caused by taking water procedures.
Aspirin in daily dose 50-250 mg, as a rule, eliminates microcirculation disorders. Taking this drug or other antiplatelet agents with therapeutic or for preventive purposes Recommended for all IP patients.
Unfortunately, there is currently no effective treatment for anemic stage III PV. Therapy is limited to palliatives. Anemic and hemorrhagic syndrome is corrected by transfusions of blood components. The effectiveness of hematopoietic stem cell transplantation in patients with PV in the stage of myelofibrosis with splenomegaly and pancytopenia and transformation into acute leukemia or MDS has been reported. The three-year survival rate of patients after transplantation was 64%.
Forecast. Despite the long and in some cases favorable course, IP is serious illness and is fraught with fatal complications that shorten the life expectancy of patients. Most common cause The deaths of patients are thrombosis and embolism (30-40%). In 20-50% of patients in the stage of postpolycythaemic myelofibrosis ( Stage III IP) transformation occurs into acute leukemia, which has an unfavorable prognosis - three-year survival rate is only 30%.
Polycythemia is a disease that can be assumed just by looking at the patient's face. And if you also carry out the necessary blood test, then there will be no doubt at all. In reference books it can also be found under other names: erythremia and Vaquez disease.
Redness of the face is quite common and there is always an explanation for this. In addition, it is short-term and does not last long. Various reasons can cause sudden redness face: fever, increased blood pressure, recent sunburn, awkward situation, and emotionally labile people generally tend to blush often, even if those around them do not see any prerequisites for this.
Polycythemia is different. Here The redness is persistent, not transient, evenly distributed throughout the face. The color of excessively “healthy” plethora is rich, bright cherry.
Polycythemia vera (erythremia, Vaquez disease) belongs to the group of hemoblastoses (erythrocytosis) or chronic ones with a benign course. The disease is characterized by the proliferation of all three germs of hematopoiesis with a significant advantage of the erythrocyte and megakaryocytic, due to which there is an increase not only in the number of red blood cells– , but also the rest of the blood cells that originate from these sprouts, where source tumor process are the affected myelopoiesis precursor cells. They are the ones who begin uncontrolled proliferation and differentiation into mature forms of red blood cells.

The ones that suffer the most under such conditions are immature red blood cells, which are hypersensitive to erythropoietin even in small doses. For polycythemia At the same time, an increase in leukocytes of the granulocytic series is observed(primarily band and neutrophils)and platelets. Cells of the lymphoid series, which include lymphocytes, are not affected by the pathological process, since they come from a different germ and have a different path of reproduction and maturation.
Erythremia is not to say that it occurs all the time, but in a town of 25 thousand people there are a couple of people, and for some reason this disease “loves” men aged 60 or so more, although anyone can encounter such a pathology age. True, for newborns and children younger age polycythemia vera is absolutely uncharacteristic, therefore if erythremia is detected in a child, then most likely she will carry secondary character and be a symptom and consequence of another disease (toxic dyspepsia, stress erythrocytosis).
For many people, the disease classified as leukemia (and it does not matter: acute or chronic) is primarily associated with blood cancer. Here it is interesting to figure out: is it cancer or not? In this case, it would be more expedient, clearer and more correct to talk about the malignancy or benignity of polycythemia vera in order to determine the boundary between “good” and “evil”. But, since the word “cancer” refers to tumors from epithelial tissues, then in in this case this term is inappropriate because this tumor comes from hematopoietic tissue.
Vaquez disease refers to malignant tumors , but is characterized by high cell differentiation. The course of the disease is long and chronic, for the time being qualified as benign. However, such a course can last only up to a certain point, and then with the right and timely treatment, but after some period of time, when significant changes occur in erythropoiesis, the disease turns into acute form and acquires more “evil” traits and manifestations. This is what it is like – true polycythemia, the prognosis of which will depend entirely on how quickly it progresses.
 Any patient suffering from erythremia sooner or later asks the question: “Why did this “disease” happen to me?” Searching for the reason of many pathological conditions, as a rule, is useful and gives certain results, increases the effectiveness of treatment and promotes recovery. But not in the case of polycythemia.
Any patient suffering from erythremia sooner or later asks the question: “Why did this “disease” happen to me?” Searching for the reason of many pathological conditions, as a rule, is useful and gives certain results, increases the effectiveness of treatment and promotes recovery. But not in the case of polycythemia.
The causes of the disease can only be assumed, but not stated unambiguously. There can only be one clue for a doctor to find out the origin of the disease - genetic abnormalities. However, the pathological gene has not yet been found, so the exact localization of the defect has not yet been determined. There are, however, suggestions that Vaquez disease may be associated with trisomy 8 and 9 pairs (47 chromosomes) or another disorder of the chromosomal apparatus, for example, loss of a section (deletion) of the long arm C5, C20, but these are still guesses, although based on conclusions of scientific research.
If there is nothing to say about the causes of polycythemia, then a lot can be said about the clinical manifestations. They are bright and varied, since already from the 2nd stage of development of the disease, literally all organs are involved in the process. The patient’s subjective sensations are of a general nature:
Complaints characteristic of this disease and characterized by it:
It's obvious that cause all these complaints - microcirculation disorder.

As further development The disease develops more and more new symptoms:
Due to the fact that for polycythemia in the first stages it is typical asymptomatic , the above manifestations do not occur in one day, but accumulate gradually and over a long period of time; in the development of the disease, it is customary to distinguish 3 stages.
Initial stage. The patient's condition is satisfactory, the symptoms are moderate, the duration of the stage is about 5 years.
Stage of advanced clinical manifestations. It takes place in two stages:
II A – occurs without myeloid metaplasia of the spleen, subjective and objective symptoms of erythremia are present, the duration of the period is 10-15 years;
II B – myeloid metaplasia of the spleen appears. This stage is characterized by a clear picture of the disease, the symptoms are pronounced, the liver and spleen are significantly enlarged.
Terminal stage, having all the signs of a malignant process. The patient’s complaints are varied, “everything hurts, everything is wrong.” At this stage, cells lose the ability to differentiate, thereby creating a substrate for leukemia, which replaces chronic erythremia, or rather, it turns into acute leukemia.
The terminal stage is characterized by a particularly severe course (hemorrhagic syndrome, rupture of the spleen, infectious and inflammatory processes that cannot be treated due to profound immunodeficiency). Usually it soon ends in death.
Thus, the life expectancy for polycythemia is 15-20 years, which may not be bad, especially considering that the disease can strike after 60. This means that there is some prospect of living up to 80 years. However, the prognosis of the disease still depends most on its outcome, that is, on what form of leukemia erythremia transforms into at stage III (chronic myeloid leukemia, myelofibrosis, acute leukemia).
 The diagnosis of polycythemia vera is primarily based on laboratory data, measuring the following indicators:
The diagnosis of polycythemia vera is primarily based on laboratory data, measuring the following indicators:
Morphologically, red blood cells do not always change and often remain normal, but in some cases, erythremia can be observed anisocytosis(red blood cells of different sizes). The severity and prognosis of the disease with polycythemia in a general blood test is indicated by platelets (the more of them, the more severe the course of the disease);
In addition to hematological parameters, to establish a diagnosis of polycythemia vera, the patient is referred for an ultrasound examination of the abdominal organs (enlarged liver and spleen).
And then the patient awaits treatment in the hematology department, where the tactics are determined clinical manifestations, hematological parameters and stage of the disease. IN therapeutic measures Erythremia usually includes:
The treatment regimen for erythremia is prescribed by the doctor individually for each case, so our task is only to briefly introduce the reader to the drugs used to treat Vaquez disease.
A significant role in the treatment of polycythemia is given to the work regime (reducing physical activity), rest and nutrition. IN initial stage illness, when the symptoms are not yet expressed or are weakly manifested, the patient is assigned to table No. 15 (general), albeit with some reservations. The patient is not recommended to consume foods that enhance hematopoiesis.(liver, for example) and suggest revising the diet, giving preference to dairy and plant products.
 In the second stage of the disease, the patient is prescribed table No. 6, which corresponds to the diet for gout and limits or completely excludes fish and meat dishes, legumes and sorrel. After being discharged from the hospital, the patient must adhere to the recommendations given by the doctor during outpatient observation or treatment.
In the second stage of the disease, the patient is prescribed table No. 6, which corresponds to the diet for gout and limits or completely excludes fish and meat dishes, legumes and sorrel. After being discharged from the hospital, the patient must adhere to the recommendations given by the doctor during outpatient observation or treatment.
Question: “Is it possible to treat folk remedies? sounds with the same frequency for all diseases. Erythremia is no exception. However, as already noted, the course of the disease and the patient’s life expectancy depend entirely on timely treatment, the goal of which is to achieve a long and stable remission and delay the third stage for as long as possible.
During the period of lull in the pathological process, the patient must still remember that the disease can return at any time, so he must discuss his life without an exacerbation with the attending physician with whom he is being observed, periodically take tests and undergo examinations.
Treatment of blood diseases with folk remedies should not be generalized, and if there are many recipes for increasing hemoglobin levels or for, this does not mean at all that they are suitable for the treatment of polycythemia, from which, in general, medicinal herbs not found yet. Vaquez's disease is a delicate matter, and in order to control the function of the bone marrow and thus influence the hematopoietic system, you need to have objective data that can be assessed by a person with certain knowledge, that is, the attending physician.
In conclusion, I would like to say a few words to the readers about relative erythremia, which cannot be confused with the true one, since relative erythrocytosis can occur against the background of many somatic diseases and end successfully when the disease is cured. In addition, erythrocytosis as a symptom can accompany prolonged vomiting, diarrhea, burn disease and hyperhidrosis. IN similar cases erythrocytosis is a temporary phenomenon and is associated primarily with dehydration of the body, when the amount of circulating plasma, which consists of 90% water, decreases.
Polycythemia vera (erythremia, Vaquez disease) refers to myeoproliferative diseases, during which, when a stem cell is damaged, proliferation of its three sprouts occurs. There is an increased formation of red blood cells, and to a lesser extent - leukocytes and platelets. Almost always with this disease, myeloid metaplasia of the spleen is observed.
The number of cases of erythremia is 1 per 100 thousand of the population, and this pathology occurs somewhat more often in men than in women. Vaquez disease is more common among people over 60 years of age than in other age groups.
The basis of the disease is the transformation of a hematopoietic stem cell, which gives the right to consider Vaquez disease as a clonal neoplastic disease.
Pluripotent cells undergo malignant transformation stem cell, which leads to the involvement of all three hematopoiesis in the process. The cells of the erythroid lineage achieve the greatest proliferation. Since this occurs in the absence of erythropoietin, this sign is specific for erythremia.
A characteristic feature of Vaquez's disease is also accumulations of polymorphic megakaryocytes.
With an increase in the mass of circulating red blood cells, the hematocrit increases and the blood becomes more viscous. The combination of these factors together with thrombocytosis leads to impaired microcirculation and frequent thromboembolic complications. In parallel, myeloid metaplasia of the spleen develops.
The disease occurs in 4 stages, reflecting pathological processes occurring in the bone marrow and spleen.
In the first stage of the disease erythrocytosis is noted, and panmyelosis is observed in the bone marrow. The durability of this steel is up to 5 years.
The disease at this stage is accompanied by plethora, acrocyanosis, burning pains and paresthesia in the fingertips. Some patients report the appearance skin itching after washing. An increase in the volume of circulating red blood cells leads to the appearance of arterial hypertension in patients who did not complain of this symptom before the onset of the disease, or aggravation of an existing one. hypertension, which is difficult to treat with traditional antihypertensive drugs. Symptoms of coronary heart disease and cerebral atherosclerosis become more pronounced.
Since the disease develops gradually, it takes 2 to 4 years from its onset to diagnosis.
Second A stage of Vaquez disease called erythremic and lasts from 10 to 15 years. In addition to erythrocytosis, an increase in the content of neutrophils with a shift to the left is observed. The picture of the bone marrow at this stage of the disease is characterized by total three-line hyperplasia with pronounced megakaryocytosis. Myeloid metaplasia of the spleen is still absent, but there is splenomegaly caused by increased sequestration of red blood cells and platelets.
This stage of the disease is accompanied by more severe and more frequent vascular complications. The risk of developing thrombotic complications is higher in elderly patients with a history of previous thrombosis.
The most frequently thrombosed blood vessels in the brain are coronary arteries and vessels supplying the abdominal organs.
Often with true polycythymia, despite increased amount platelets, hemorrhagic syndrome is observed, manifested by nosebleeds and heavy blood loss after tooth extraction. This occurs due to the slow conversion of fibrinogen to fibrin and impaired blood clot retraction.
Visceral complications of Vaquez disease can include erosions and ulcers of the stomach and duodenum. They are a consequence of impaired blood flow and trophism of the gastric and intestinal mucosa.
Second B stage of erythremia differs by the addition of myeloid metaplasia of the spleen. Splenomegaly becomes more pronounced. In the peripheral blood, leukocytosis is observed, a shift of the formula to the left occurs to juvenile forms. The bone marrow is characterized by panmyelosis. Some stabilization of the condition of patients at this stage of the disease may be due to a decrease in the level of red blood cells due to their increased destruction by the spleen.
Stage three of Vaquez's disease was called anemic. In addition to the phenomenon of anemia, thrombocytopenia and pancytopenia are observed in the peripheral blood. In parallel, splenomegaly and cachexia increase.
Criteria allowing diagnose polycythemia vera, are divided into main and additional.
The first includes an increase in the mass of circulating red blood cells with normal saturation arterial blood oxygen, as well as splenomegaly.
Additional symptoms include thrombocytosis and leukocytosis in the absence of signs of infection, increased alkaline phosphatase activity above 100 units, and high levels of vitamin B12.
Currently great importance In the diagnosis of erythremia, the bone marrow picture is attached.
To prevent thrombosis, aspirin is prescribed in a dose of 50 to 250 mg per day.
Often, with the correction of microcirculation, the symptoms of itching after a hot bath also disappear. However, in some cases, to eliminate this symptom it is necessary to resort to ultraviolet irradiation of autologous blood.
To reduce the number of red blood cells, bloodletting is used, which must be done with extreme caution. Frequent bloodletting can lead to iron deficiency, which does not require correction without signs of tissue deficiency of this element.
Severe erythremia requires use medicinal methods decrease in the number of red blood cells. For this purpose, hydroxyurea drugs, alpha-interferon, and myelosan are used.
Despite the fact that the disease takes a long time and is relatively benign, fatal outcomes with Vaquez disease are possible due to developing complications diseases. Most often, the patient’s life is threatened by thrombosis and thromboembolism, as well as transformation of the disease into acute leukemia.
Preventive measures for erythremia are aimed at early detection diseases and preventing the development of complications of the disease.
Polycythemia vera (erythremia, Vaquez disease or primary polycythemia) is a progressive leukemia. malignant disease, which is associated with hyperplasia of cellular elements of the bone marrow (myeloproliferation). The pathological process primarily affects the erythroblastic germ, so an excess number of red blood cells is detected in the blood. There is also an increase in the number neutrophil leukocytes and platelets.
| ICD-10 | D45 |
|---|---|
| ICD-9 | 238.4 |
| ICD-O | M9950/3 |
| MedlinePlus | 000589 |
| MeSH | D011087 |
An increased number of red blood cells increases blood viscosity, increases its mass, causes a slowdown in blood flow in the vessels and the formation of blood clots. As a result, patients develop impaired blood supply and hypoxia.
Polycythemia vera was first described in 1892 by the French physician and cardiologist Vaquez. Vaquez suggested that the hepatosplenomegaly and erythrocytosis detected in his patient arose as a result of increased proliferation of hematopoietic cells, and identified erythremia as a separate nosological form.
In 1903, W. Osler used the term “Vaquez disease” to describe patients with splenomegaly (enlarged spleen) and severe erythrocytosis and gave a detailed description of the disease.
Turk (W. Turk) in 1902-1904 suggested that in this disease the disorder of hematopoiesis is hyperplastic in nature, and called the disease erythremia by analogy with leukemia.
The clonal neoplastic nature of myeloproliferation, which is observed in polycythemia, was proven in 1980 by P. J. Fialkov. He discovered one type of enzyme, glucose-6-phosphate dehydrogenase, in red blood cells, granulocytes and platelets. In addition, both types of this enzyme were detected in the lymphocytes of two patients heterozygous for this enzyme. Thanks to Fialkov's research, it became clear that the target of the neoplastic process is the precursor cell of myelopoiesis.
In 1980, a number of researchers managed to separate the neoplastic clone from normal cells. It has been experimentally proven that in polycythemia a population of erythroid committed precursors is formed that have pathologically high sensitivity even to a small amount of erythropoietin (kidney hormone). According to scientists, this contributes to increased formation of red blood cells in polycythemia vera.
In 1981, L. D. Sidorova and co-authors conducted studies that made it possible to detect qualitative and quantitative changes in the platelet component of hemostasis, which play a major role in the development of hemorrhagic and thrombotic complications in polycythemia.
Polycythemia vera is detected mainly in older people, but can be observed in young people and children. In young people, the disease is more severe. The average age of patients varies from 50 to 70 years. The average age of those who become ill for the first time is gradually increasing (in 1912 it was 44 years, and in 1964 - 60 years). The number of patients under 40 years of age is about 5%, and erythremia in children and patients under 20 years of age is detected in 0.1% of all cases of the disease.
Erythremia is slightly less common in women than in men (1: 1.2-1.5).
It is the most common disease in the group of chronic myeloproliferative diseases. It is quite rare - according to various sources, from 5 to 29 cases per 100,000 population.
There is scattered data on the influence of racial factors (above average among Jews and below average among representatives Negroid race), however at the moment this assumption has not been confirmed.
Polycythemia vera is divided into:
An absolute increase in erythrocyte mass is observed in all patients, but only in 2/3 the number of leukocytes and platelets also increases.
The causes of polycythemia vera have not been definitively established. Does not currently exist unified theory, which would explain the occurrence of hemoblastoses (blood tumors), to which this disease belongs.
Based on epidemiological observations, a theory was put forward about the connection of erythremia with the transformation of stem cells, which occurs under the influence of gene mutations.
It has been established that most patients have a mutation in the enzyme Janus kinase-tyrosine kinase, synthesized in the liver, which is involved in the transcription of certain genes by phosphorylating many tyrosines in the cytoplasmic part of the receptors.
The most common mutation, discovered in 2005, is in exon 14 JAK2V617F (detected in 96% of all cases of the disease). In 2% of cases, the mutation affects exon 12 of the JAK2 gene.
Patients with polycythemia vera also have:
Elderly patients with a high JAK2V617F allelic load are characterized by increased level hemoglobin, leukocytosis and thrombocytopenia.
With a mutation of the JAK2 gene in exon 12, erythremia is accompanied by a subnormal serum level of the hormone erythropoietin. Patients with this mutation are younger.
In polycythemia vera, mutations of TET2, IDH, ASXL1, DNMT3A, etc. are also often detected, but they are pathogenetic significance has not yet been studied.
Differences in survival of patients with different types no mutations were detected.
As a result of molecular genetic disorders, the JAK-STAT signaling pathway is activated, which is manifested by proliferation (cell production) of the myeloid lineage. At the same time, proliferation and an increase in the number of red blood cells in the peripheral blood increase (an increase in the number of leukocytes and platelets is also possible).
The identified mutations are inherited in an autosomal recessive manner.
There is also a hypothesis according to which the cause of erythremia may be viruses (15 types of such viruses have been identified), which, in the presence of predisposing factors and weakened immunity, penetrate into immature bone marrow cells or lymph nodes. Cells affected by the virus begin to actively divide instead of maturing, thus starting the pathological process.
Factors that provoke the disease include:
Secondary erythremia develops under the influence of favorable factors when:
The pathogenesis of polycythemia vera is associated with a disruption of the process of hematopoiesis (hematopoiesis) at the level of the progenitor cell. Hematopoiesis acquires the unlimited proliferation of progenitor cells characteristic of a tumor, the descendants of which form a specialized phenotype in all hematopoietic lineages.
Polycythemia vera is characterized by the formation of erythroid colonies in the absence of exogenous erythropoietin (the appearance of endogenous erythropoietin-independent colonies is a sign that distinguishes erythremia from secondary erythrocytosis).
The formation of erythroid colonies indicates a disruption in the implementation of regulatory signals that the myeloid cell receives from the external environment.
The basis of the pathogenesis of polycythemia vera is defects in genes encoding proteins that are responsible for maintaining myelopoiesis within the normal range.
A decrease in oxygen concentration in the blood causes a reaction in the interstitial cells of the kidneys that synthesize erythropoietin. The process occurring in interstitial cells concerns the work of many genes. The main regulation of this process is carried out by factor-1 (HIF-1), which is a heterodimeric protein consisting of two subunits (HIF-1alpha and HIF-1beta).
If the oxygen concentration in the blood is within normal limits, proline residues (the heterocyclic amino acid of the freely existing HIF-1 molecule) are hydroxylated under the influence of the regulatory enzyme PHD2 (molecular oxygen sensor). Thanks to hydroxylation, the HIF-1 subunit acquires the ability to bind to the VHL protein, which provides tumor prevention.
The VHL protein forms a complex with a number of E3 ubiquitin ligase proteins, which, after forming covalent bonds with other proteins, are sent to the proteasome and destroyed there.
During hypoxia, hydroxylation of the HIF-1 molecule does not occur; the subunits of this protein combine and form the heterodimeric HIF-1 protein, which travels from the cytoplasm to the nucleus. Once in the nucleus, the protein binds to special DNA sequences in the promoter regions of genes (the conversion of genes into protein or RNA is induced by hypoxia). As a result of these transformations, erythropoietin is released into the bloodstream by the interstitial cells of the kidneys.
By myelopoiesis precursor cells, the genetic program embedded in them is carried out as a result of the stimulating effect of cytokines (these small peptide control (signal) molecules bind to the corresponding receptors on the surface of the precursor cells).
When erythropoietin binds to the erythropoietin receptor EPO-R, dimerization of this receptor occurs, which activates Jak2, a kinase associated with the intracellular domains of EPO-R.
Jak2 kinase is responsible for signal transmission from erythropoietin, thrombopoietin and G-CSF (granulocyte colony-stimulating factor).
Due to the activation of Jak2-kinase, phospholation of a number of cytoplasmic target proteins occurs, which includes adapter proteins of the STAT family.
Erythremia was detected in 30% of patients with constitutive activation of the STAT3 gene.
Also, with erythremia, in some cases, a reduced level of expression of the thrombopoietin receptor MPL is detected, which is compensatory in nature. The reduction in MPL expression is secondary and is caused by a genetic defect responsible for the development of polycythemia vera.
A decrease in degradation and an increase in the level of the HIF-1 factor is caused by defects in the VHL gene (for example, representatives of the population of Chuvashia are characterized by a homozygous mutation 598C>T of this gene).
Polycythemia vera can be caused by abnormalities of chromosome 9, but the most common is a deletion of the long arm of chromosome 20.
In 2005, a point mutation in exon 14 of the Jak2 kinase gene (mutation JAK2V617F) was identified, which causes the replacement of the amino acid valine with phenylalanine in the pseudokinase domain JH2 of the JAK2 protein at position 617.
The JAK2V617F mutation in hematopoietic precursor cells in erythremia is presented in a homozygous form (the formation of the homozygous form is affected by mitotic recombination and duplication of the mutant allele).
When JAK2V617F and STAT5 are active, the level of reactive oxygen species increases, resulting in a transition of the cell cycle from G1 to S phase. Adapter protein STAT5 and active forms oxygen transmit a regulatory signal from JAK2V617F to the cyclin D2 and p27kip genes, which causes an accelerated transition of the cell cycle from the G1 to S phase. As a result, the proliferation of erythroid cells that carry a mutant form of the JAK2 gene increases.
In JAK2V617F-positive patients, this mutation is detected in myeloid cells, B- and T-lymphocytes and natural killer cells, which proves the proliferative advantage of defective cells compared to the norm.
Polycythemia vera in most cases is characterized by a fairly low ratio of mutant to normal allele in mature myeloid cells and early precursors. In the presence of clonal dominance, patients experience more severe clinical picture compared to patients without this defect.
Symptoms of polycythemia vera are associated with excess production of red blood cells, which increase blood viscosity. In most patients, the level of platelets, which cause vascular thrombosis, also increases.
The disease develops very slowly and initial stage is asymptomatic.
For more later stages Polycythemia vera manifests itself:
Plethoric syndrome is accompanied by:
Myeloproliferative syndrome manifests itself:
Varicose veins are also observed, especially noticeable in the neck area, Cooperman's sign (change in color of the soft palate with normal coloration of the hard palate), duodenal ulcer and, in some cases, stomach, bleeding of the gums and esophagus, and increased uric acid levels. The development of heart failure and cardiosclerosis is possible.
Polycythemia vera is characterized by three stages of development:
Erythremia is diagnosed based on:
A coagulogram, iron metabolism studies are also performed, and the level of erythropoietin in the blood serum is determined.
Since chronic erythremia is accompanied by an enlargement of the liver and spleen, an ultrasound of the internal organs is performed. Ultrasound also detects the presence of hemorrhages.
To assess the extent of the tumor process, SCT (spiral CT scan) and MRI (magnetic resonance imaging).
To identify genetic abnormalities, a molecular genetic study of peripheral blood is performed.
The goals of treatment for polycythemia vera are:
Erythremia is treated with:
To prevent thrombosis, anticoagulants are used (usually acetylsalicylic acid is prescribed at 40-325 mg/day).
Nutrition for erythremia must meet the requirements of the treatment table according to Pevzner No. 6 (the amount of protein foods is reduced, red fruits and vegetables and foods containing dyes are excluded).

In 2018, the Nativity Fast will begin on November 28. During this period, Orthodox believers prepare to celebrate Christmas...

Starting a family is the dream of most women. They want to have a loving husband and a bunch of kids. But it's not always a relationship...

This article contains: the most powerful prayer for divorce - information taken from all over the world, the electronic network and...

Information site about icons, prayers, Orthodox traditions. Prayer for scandals and quarrels in the family, with husband, with children...

What would New Year be without champagne, tangerines, Olivier, aspic and everyone’s favorite “Herring under a fur coat”. With the last one...

Let's prepare the necessary ingredients for the cookies. The first thing to do is put the water to boil. We need...

Is it possible to register an employee for the position of financial director - chief accountant? The chief accountant claims...

The head of a small business can easily manage the budget independently. CHECKED! If you manage...

The creation of new projects involves a preliminary economic justification for their feasibility, subsequent...
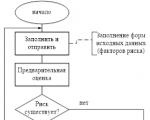
Reporting is generated by the RM, is agreed upon (approved) by the Risk Committee under the Management Board and transmitted to...

At the edge of a large, very large meadow, on a long emerald blade of grass lived a tiny Ladybug. God's little...

Nowadays, it is quite common for people to turn to the stars. With the help of a horoscope a person can find out...

Business or friendly. If you were pursuing a stranger, it means your level of trust in the female sex...

according to Freud's dream book If you dreamed about how you were fishing, it means that in real life you can hardly switch off...

Starting a family is the dream of most women. They want to have a loving husband and a bunch of kids. But it's not always...

This article contains: the most powerful prayer for divorce - information taken from all over the world, electronic...